
Photovoltaic poly(rod-coil) polymers based on benzodithiophene-centred A–D–A type conjugated segments and dicarboxylate-linked alkyl non-conjugated segments†
Abstract
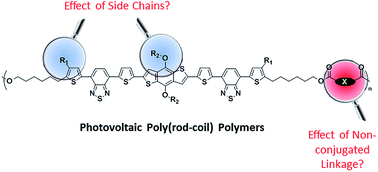
Different from the well-studied photovoltaic conjugated polymers and small molecular compounds, poly(rod-coil) polymers are emerging as a new class of photovoltaic materials. Since they are composed of definite conjugated and non-conjugated segments in an alternative fashion, this kind of material is expected to merge the merits from both small molecular compounds and conjugated polymers. Based on benzodithiophene-centered acceptor–donor–acceptor (A–D–A) conjugated segments and dicarboxylate-linked alkyl non-conjugated segments, this study has newly designed and synthesized two poly(rod-coil) polymers. Together with three previously reported analogues, these polymers have been systematically investigated for their photovoltaic performances, with special attention paid to the effect of the dicarboxylate linking unit in non-conjugated segments and the alkyl side chains on rigid conjugated segments. It was found that the former factor has a small influence, while the latter has a significant impact on most film-related properties of the material, including film absorption spectrum, frontier orbital energy levels, bandgap, microstructure and morphology of pristine and photovoltaic blend films, as well as hole mobility. After optimization, bulk heterojunction organic solar cells based on this series of polymers reported power conversion efficiencies in range of 0.4-1.09%.
 Please wait while we load your content...
Please wait while we load your content...

