DOI:
10.1039/C6RA01145D
(Paper)
RSC Adv., 2016,
6, 23252-23259
Sequestration of naturally abundant seawater calcium and magnesium to enhance the adsorption capacity of bentonite toward environmental phosphate
Received
14th January 2016
, Accepted 20th February 2016
First published on 22nd February 2016
Abstract
The consideration of the water energy nexus inspires environmental engineers to pursue a more sustainable remediation method for pollution control and resource recovery. In this study, a cost-effective strategy is proposed to enhance the adsorption capacity of bentonite toward environmental phosphate by sequestrating naturally abundant seawater calcium and magnesium. By adopting this method, the adsorption capacity of modified bentonite toward phosphate is enhanced by two orders of magnitude (11.45 vs. 0.11 mg g−1). This achievement can be realized as a result of the intercalated seawater Ca2+/Mg2+ to screen the negative charge of aluminum phyllosilicate sheets of bentonite. The results from an additional SEM/EDX analysis and FTIR characterization imply that adsorbed phosphate is strongly associated with the intercalated Ca2+/Mg2+ in a configuration similar to that of hydroxyapatite. Moreover, our results demonstrate that the acquisition of ocean resources would be a promising and green strategy for the perspective of environmental remediation.
1. Introduction
Phosphate is an essential nutrient responsible for spurring eutrophication and algae blooms, which lead to serious environmental concerns with regard to the quality of streams, lakes and other water bodies. To avoid the occurrence of eutrophication, the concentration of phosphate in these water bodies should be controlled below 0.05 mg L−1. Unlike industrial wastewater, phosphate concentration in these phosphate-rich natural water bodies usually lies on the level of several parts per million. This means that the cost of removing phosphate from these aquatic systems by chemical methods such as precipitation would be significant. In this case, the utilization of adsorbents as an alternative would be a reasonable solution. In addition, consideration of the water energy nexus inspires environmental engineers to pursue a more sustainable water treatment process. To respond to this challenge, a simple and less energy-consuming strategy to prepare an environmentally friendly adsorbent is highly desired.
Several studies have been carried out to prepare inexpensive and effective adsorbents to remove environmental phosphate from natural water bodies. Among them, the low-cost clay could be a promising adsorbent owing to its abundance worldwide. Despite the advantage of its low cost, natural clay usually exhibits very low phosphate adsorption capacity. This is because the surface of aluminum phyllosilicate sheets of clays always possesses permanent negative charges owing to the isomorphous substitution effect. This explains the observations that soil samples that contain low clay content (<10%) usually have high phosphate trapping capability owing to their internal Al- and Fe oxide impurity.1,2 Moreover, low phosphate leaching is often found in sandy soils that contain high Al2O3 content.3 Based on these field observations, surface modification with high oxidation state elements is often applied to reduce or compensate for the permanent negative charges on clay surfaces. For example, Al2O3 coated clay exhibits a phosphate adsorption capacity two times higher than that of the unmodified clay.4 The lanthanum (La)-modified bentonite clay (Phoslock) was demonstrated to possess high phosphate adsorption capacity in a wide salinity range.5 Intercalation with polyhydroxyl iron through cation exchange reaction could effectively reduce the surface charge of bentonite and thus enhance the adsorption capacity of modified bentonite toward anionic pollutants.6 Similar results are also observed in clay samples intercalated with cation ionic surfactants such as hexadecylammonium bromide.7,8 Zirconium is another high-oxidation-state element used to modify clays to improve the phosphate adsorption capacity.9 Triethanolamine-grafted kaolinite and Fe-intercalated bentonite have also been demonstrated to exhibit at least three times higher phosphate removal capacity than their parent clays.10,11
In addition to the usage of Al, Fe, La and Zr, alkaline earth elements such as Ca and Mg are also considered for the modification of bentonite. By the intercalation of CaO into gastropod shell clays, high phosphate adsorption capacity of modified gastropod shell clay could be achieved.12 Similarly, through the slow pyrolysis of Mg-enriched tomato tissues in an oxygen-deficient environment, engineered biochar embedded with nanoscale Mg(OH)2 and MgO is found to have a high phosphate adsorption capacity because these nanoparticles serve as the main adsorption sites for phosphate accommodation.13 These interesting results clearly indicate that the appropriate acquisition of naturally abundant alkaline earth elements would be an environmentally friendly route to prepare high-performance adsorbents for the removal of phosphate.14 In this study, we demonstrate the acquisition of naturally abundant seawater magnesium and calcium by intercalating them into the interlayer space of bentonite. After sintering at 600 °C to convert intercalated Mg2+/Ca2+ to MgO/CaO, the permanent negative charge of aluminum phyllosilicate sheets of bentonite is effectively compensated. Further, intercalated MgO/CaO would bridge the aqueous phosphate to negatively charged bentonite surfaces.15 This property allows the modified bentonite to achieve an adsorption capacity two orders of magnitude higher than that of raw bentonite. The success of sequestration of seawater Ca/Mg means that the environmental impact due to Ca/Mg mineral exploitation could be greatly reduced, whereas as-obtained adsorbents would be cost effective for the application of environment remediation. Our proposed modification would therefore be an important reference for the preparation of green adsorbents for the removal of other anionic contaminants such as arsenic and antimony in the future.
2. Experimental
2.1. Sequestration of seawater calcium/magnesium
The seawater used in this study was collected from the coast area near Ningbo, Zhejiang province, China. Collected seawater was first filtrated using a microfiltration membrane (0.22 μm pore size) to remove suspensions. The modification of bentonite was conducted by immersing Na-type MX-80 bentonite in the purified seawater at room temperature, and the Mg2+- and Ca2+-loaded bentonite was then sintered at 600 °C. Briefly, 20.0 g of bentonite and 1000 mL of seawater were mixed in a 2.0 L plastic container and stirred at 300 rpm for 360 min at room temperature. At the end of the mixing, the Mg2+- and Ca2+-loaded bentonite was collected by filtration and dried at 120 °C. At this stage, loaded Mg2+ and Ca2+ were presented in the form of hydrated Mg and Ca (Mg/Ca coordinated with water molecules), respectively. The last stage was to sinter the dried bentonite powders in air at 600 °C for 1 h, which converted the loaded hydrated Mg and Ca to MgO and CaO, respectively. The MgO/CaO-loaded bentonite was labeled as BS600, whereas the raw bentonite that was subjected through the identical sintering process was prepared for the reference set and labeled as B600. For characterizing interlayer environment of BS600, both Ca- and Mg-saturated bentonite were also prepared with the same solid/liquid ratio as mentioned above.16 This was done by dispersing MX-80 bentonite in 1.0 M NaCl solution for one hour and collecting them by centrifuge; this Na exchange process was repeated for three times. This step was conducted to replace all exchangeable interlayer cations by Na+ solution. These Na-saturated MX-80 bentonite samples were then dispersed in 1.0 M Mg/Ca solution and centrifuged three times. After Mg/Ca exchange, Mg/Ca saturated MX-80 bentonite samples were also subjected to sintering following an identical preparation procedure to that mentioned above. These samples were labeled as BCa600 and BMg600 in the following context, respectively. This relatively complicated Mg/Ca exchange procedure is very important since it minimizes the full width at half maximum in d001 diffraction that benefits the explicit revelation of the interlayer environment of the treated bentonite.16
2.2. Adsorption experiments
All adsorption experiments were carried out in triplicate in a 500 mL conical flask by dispersing 200 mg of B600/BS600 in 200 mL of 10 mM NaCl solution (solid/liquid ratio = 1/1000) at room temperature. The mixtures were allowed to equilibrate for 24 h under continuous mixing using a temperature-controlled thermostatic shaker. To evaluate the performance of as-prepared adsorbents, the adsorption experiments were conducted with the initial phosphate concentration maintained at 5 ppm (0.05 mM) as a function of pH. The initial pH of the phosphate solutions was adjusted using either 0.1 M NaOH or HCl solution, whereas the equilibrated pH was recorded using a digital pH meter (UB-7, Denver, USA). For isotherm experiments, the equilibrated pH of the phosphate solutions was at pH 7.5 ± 0.3 where the initial phosphate concentrations were in the range from 0.1–50 ppm. At the end of adsorption experiments, the mixture was centrifuged, the supernatant was collected, and the equilibrated phosphate concentration was determined using inductively coupled plasma optical emission spectrometry (ICP-OES 5100 DV, Agilent). The wet clay pastes were also collected for subsequent SEM/EDX and FTIR analysis. The removal efficiency (R) and the amount of adsorbed phosphate on the surface of B600 and BS600 (qe, mg g−1) were calculated using the following equations:| |
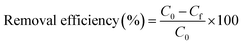 | (1) |
| |
 | (2) |
where V (L) is volume of the solution, m (g) is the mass of the added B600/BS600, and C0 and Cf (mg L−1) are the initial and equilibrated phosphate concentrations, respectively. The as-obtained adsorption isotherms were fitted using the Langmuir (eqn (3)) and Freundlich model (eqn (4)) as shown below:| |
 | (3) |
where Ce is the equilibrium concentration of phosphate in the solution (mg L−1); qm and qmax are the amount of adsorbed phosphate and the maximum adsorption capacity of the adsorbent (mg g−1), respectively; kd is the Langmuir adsorption equilibrium constant (L mg−1); K is the Freundlich constant (mg g−1) and n is a constant describing adsorption linearity.
2.3. Determination of the points of zero charge of the B600 and BS600
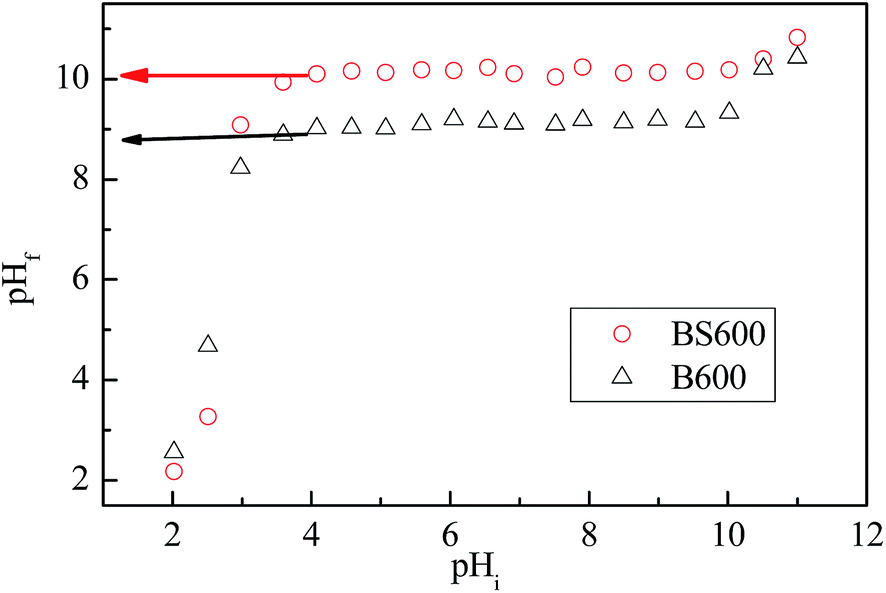
Considering the large particle size of bentonite that is extremely susceptible to precipitation during zeta potential measurement, the points of zero charge of B600 and BS600 were alternatively determined according to following procedure.17 First, the initial pH values of the NaCl (10 mM) solutions were adjusted by adding either 0.1 M HCl or NaOH solutions. One hundred milligrams of B600/BS600 was then dispersed in 100 mL of NaCl solution in a conical flask under vigorous stirring. Equilibration was allowed for 24 h at room temperature, and the equilibrated pH (pHf) was recorded. The point of zero charge of B600/BS600 was thus determined from the plot of pHf against pHi at which the curve plateau (or the curve inflexion) appears. It is worth mentioning that whereas the zeta potential measurement is a more frequently adopted method for pHzpc determination, it is severely susceptible to the precipitation effect, particularly to colloids with sizes on the micrometer scale such as bentonite particles. Accordingly, the batch equilibrium technique was selected in this study instead.
2.4. Characterization
Powder X-ray diffraction (PXRD) patterns were obtained at room temperature with an X'Pert ProMPD diffractometer (X'Pert Pro, PANalytical) using a Cu Kα radiation (λ = 0.15418 nm) beam in the 2θ range of 3–80°. The specific surface area and porous properties of B600 and BS600 were characterized using nitrogen physisorption isotherms (N2-BET, Micromeritics ASAP 2020). The texture and surface elemental composition of B600 and BS600 were examined using scanning electron microscopy equipped with an energy-dispersive X-ray spectrometer (SEM/EDX; JSM700F, JEOL). The ATR-FTIR spectra of BS600 were recorded in the wavelength range of 4000–400 cm−1 for 64 scans with a resolution of 4 cm−1 (Nicolet iS10, Thermo).
3. Results and discussion
3.1. Characterization of seawater and modified bentonite
The composition of seawater is shown in Fig. 1. While the minor mineral content in seawater might change depending on the location, the major mineral content (Na, Cl, Mg, Ca) is rather constant in which the sum of Mg and Ca content contributes approximately 5 wt%, which is consistent with the value reported in the literature.14 Importantly, because the seawater Na content is at least 10 times higher than that of Mg and Ca, one might reasonably expect that immersing Na-type bentonite in seawater would have a very limited effect on acquiring seawater Ca/Mg on the surface of bentonite. Accordingly, the acquisition of seawater Ca/Mg might fail because more Na would be exchanged to bentonite, which is identical to the nature of raw bentonite. It is true that there would be a large amount of Na intercalated into bentonite owing to its high content in seawater. For instance, given the that theoretical cation exchange capacity (CEC) of MX-80 is 760 meq. kg−1,16 this indicates that when dispersing 0.02 kg MX-80 in 1 L deionized water (the same solid/liquid ratio as we prepared BCa600 and BMg600), the maximum released amount of exchangeable interlayer cation is about 0.0152 mole Na (equals to 0.0076 mole Ca). Since the molar composition of Na and Ca in seawater (salinity = 35) is 0.469 M and 0.0103 M, respectively, the amount of released Na for MX-80 bentonite and exchanged for seawater Mg/Ca into MX-80 bentonite is likely to be insignificant (note: ICP-MS is absolutely capable of determining the change in Na, Mg, Ca concentration in seawater but determined values would be highly vulnerable to error propagation in sample preparation and analysis processes). However, the competitive adsorption (selectivity) based on thermodynamics would preferentially allow the intercalation of seawater Ca/Mg as they would neutralize and stabilize the permanent charge of aluminum phyllosilicate sheets more effectively than the alkali cations.18 Indeed, the adsorption selectivity of bentonite ensures the success of acquisition of seawater Ca/Mg, that avoids the exploitation of Ca and Mg minerals and thus greatly reduces the environmental impact resulting from the adsorbent preparation.
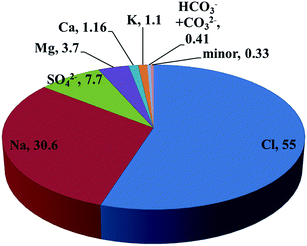 |
| | Fig. 1 The weight composition of elements in seawater adopted in this study. | |
To confirm the success of modification, the d001 diffraction peaks of dried powders of B600 and BS600 collected at the end of immersion were recorded and are shown in Fig. 2. It is noted that the d001 diffraction of BS600 shifts from 2θ = 9.24° to 2θ = 8.98°, which corresponds with the expansion of interlayer space from 9.56 Å to 9.84 Å. For the purpose of comparison, the d001 diffractions of BCa600 and BMg600 are also presented in Fig. 2a, the interlayer spaces of which are determined to be 9.84 Å and 9.70 Å, respectively. From Fig. 2a, it is clear that at the end of immersion, the majority of interlayer cations are replaced by seawater Ca, resulting in a shift of d001 diffraction to the lower diffraction angle. This is a solid piece of evidence supporting our above speculation that adsorption selectivity of bentonite ensures the success of acquisition of seawater Ca/Mg. Interestingly, although seawater contains more Mg (3.7 wt%) than Ca (1.2 wt%, Fig. 1) by approximately three times, the results from XRD analyses suggest that the modification preferentially leads to the development of Ca-type rather than Mg-type bentonite, although a small fraction of Mg-type and Na-type bentonite is concurrently noted as indicated by the broadened d001 diffraction (P2). This is because the higher hydration energy of Ca than that of Mg and thus the stabilization of the permanent charge of aluminum phyllosilicate sheets is more effective when intercalating the former than when intercalating the latter. Furthermore, at the end of sintering, the intercalated Ca cation is transformed to calcium oxide as suggested by the appearance of diffraction peaks at 32.20°, 37.35° and 53.85° (2θ) (as shown in Fig. 2b). Because CaO is known to have a high affinity to phosphate, it is reasonably anticipated that the BS600 would exhibit a high adsorption capacity to phosphate.
 |
| | Fig. 2 (a) The d001 diffraction peaks and (b) full diffraction spectra of modified bentonite samples. | |
In addition to XRD patterns, another solid piece of evidence suggesting the intercalation of seawater Mg/Ca in bentonite can be noted from the N2 adsorption/desorption isotherms as shown in Fig. 3. As a rule, calcination of clay minerals might lead to a reduction in their specific surface area; results from BET analyses suggest the difference in surface area between calcinated and raw samples is not significant (date not shown due to superimposition). This can be explained by the nonporous nature of bentonite particles and therefore the determined surface area of bentonite aggregates is mainly composed by mesopores and macropores stemming from the stacking of bentonite particles. By contrast, the surface area of the BS600 is noted to be greatly reduced by approximately 25.5% from 37.34 m2 g−1 to 27.80 m2 g−1. The reduction in the surface area can be realized owing to the loss of a large fraction of mesoporous materials (insert, Fig. 3) as indicated by the largely disappeared hysteresis loops at relative pressures P/P0 of 0.45–1.0. Similar behavior has been reported in the preparation of organoclays where the intercalated cationic surfactants block the pores of clay particles and thus the surface area of the former is much smaller than that of the latter.8 We suspect that a similar behavior has also occurred in our system, where the formation of CaO/MgO particles blocks the pores of bentonite and therefore greatly reduces the surface area of BS600.
 |
| | Fig. 3 N2 adsorption/desorption isotherms and pore size distribution (insert) of B600 and BS600. | |
Fig. 4 shows the SEM images of B600 and BS600. Compared with the sample B600 (Fig. 4a), some fine calcium oxide particles appear on the surface of BS600 (black arrows, Fig. 4b). Results from additional EDX analyses (Fig. 4c) indicate that the surface of BS600 contains a large amount of Na, Ca, Mg, K, and Cl, which is rather different from EDX spectrum of raw bentonite (Fig. 4d). This again confirms the success of the modification and demonstrates the potential of sequestrating seawater resources to prepare functional materials as a practical solution for environmental problems. According to the EDX results, the contents of CaO and MgO were estimated as 7 wt% and 2 wt% in BS600, respectively.
 |
| | Fig. 4 The SEM image of (a) B600 and (b) BS600; and the EDX spectra of (c) BS600 and (d) B600. Inserted scale bars refer to 1 μm. | |
3.2. Adsorption behavior of modified bentonite
The adsorption performance of B600 and BS600 is shown in Fig. 5. It is noted that a very limited amount of phosphate (<5%) is adsorbed by B600 in all studied pH environments (Fig. 5a). This is realized by the electrostatic repulsion between the phosphate and the negatively charged bentonite surface. In contrast, the adsorption of phosphate on BS600 is greatly enhanced to approximately 80% when pH > 4. Importantly, high phosphate adsorption (>80%) is noted even in the alkaline environment, where both BS600 and phosphate are negatively charged. This means that instead of electrostatic interactions, the high affinity between CaO and phosphate through the formation of calcium phosphate configuration is likely to be responsible for the high phosphate uptake. Results from phosphate adsorption to BCa600 and BMg600 are also shown in Fig. 5a, where a less pH-sensitive adsorption behavior is noted. This is a clear piece of evidence indicating that electrostatic interaction is not the dominant driving force responsible for the observed adsorption behavior.19–21 Similar phosphate adsorption behavior by lanthanum-modified bentonite has been reported in which the formation of rhabdophane mineral was clearly identified by using P-31 SSNMR, La-139 SSNMR and EXAFS.22 In that study, the authors suggested that water molecules also play an important role in phosphate uptake as they would be the bridge (through a configuration similar to hydrogen bonding) to connect highly charged La3+ and phosphate. This configuration could probably explain the low phosphate uptake by BS600, BCa600 and BMg600 in a low pH environment as in these conditions fewer hydrogen bonds are formed. The isotherm of phosphate adsorption to B600 and BS600 conduced at pH 7.5 ± 0.3 is shown in Fig. 5b, in which the maximum adsorption capacities (qm) of B600 and BS600 determined by the Langmuir model (solid lines) are 0.1 and 11.45 mg g−1, respectively (Table 1). As relatively greater correlation coefficients (R2) were obtained by using the Langmuir model (solid lines) than by using the Freundlich model (dash lines), this means that the phosphate uptake by both B600 and BS600 more likely follows the monolayer adsorption. Another interesting feature revealed by Fig. 5 is that Cs has a higher phosphate adsorption capacity than Mg does, which again supports our above statement. In addition to the enhanced adsorption capacity, the affinity of BS600 is also greatly improved as indicated by the greater Langmuir constant, which lies between that of BCa600 and BMg600 (kd, Table 1). It is therefore speculated that the intercalated seawater Ca/Mg not only screens/neutralizes the permeated charge of aluminum phyllosilicate sheets of bentonite but is also the adsorption site for phosphate accommodation. We further estimate the adsorption site density of these samples by dividing their maximum adsorption capacity determined from the Langmuir model by their surface area. It is noted that adsorption site density is greatly increased by approximately two orders of magnitude from 0.02 to 2.61 site per nm2, which clearly indicates the success of our proposed modification method. On the other hand, the determined adsorption site density of BS600 is found to be close to that of BCa600, this might be a piece of indirect evidence suggesting intercalated seawater Ca plays an important role in phosphate uptake. However, additional spectroscopic analysis is highly desirable to reveal the involved mechanism due to the relatively low correlation coefficients (Table 1).
 |
| | Fig. 5 (a) Effect of pH on the adsorption of phosphate and (b) the adsorption capacity of B600, BS600, BCa600 and BMg600, respectively. | |
Table 1 Best-fitting results using the Langmuir and Freundlich model, values shown in the parenthesis are the standard errors (Fig. 5b)
| |
Langmuir model |
Freundlich model |
| R2 |
kd/L mg−1 |
qmax/mg g−1 |
Site per nm2 |
R2 |
K/mg g−1 |
n |
| B600 |
0.7668 |
0.20 (0.07) |
0.11 (0.02) |
0.02 (0.01) |
0.7379 |
0.02 (0.01) |
2.39 (0.13) |
| BS600 |
0.8810 |
0.31 (0.07) |
11.45 (1.86) |
2.61 (0.42) |
0.7336 |
1.91 (0.25) |
1.88 (0.09) |
| BCa600 |
0.9032 |
0.38 (0.08) |
13.73 (1.00) |
3.12 (0.37) |
0.9798 |
5.32 (0.01) |
3.14 (0.11) |
| BMg600 |
0.9163 |
0.23 (0.02) |
7.49 (0.17) |
1.71 (0.33) |
0.6727 |
3.35 (0.23) |
5.76 (0.75) |
To evaluate the degree of neutralization by intercalated seawater Ca/Mg, the points of zero charge (pHzpc) of samples B600 and BS600 were determined using the batch equilibration technique.17 From Fig. 6, the determined pHzpc values for B600 and BS600 are 8.8 and 10.1, respectively. The increase in the point of zero charge as a result of intercalation of seawater Mg/Ca indicates a decrease in electrostatic repulsion between the bentonite surface and phosphate, which likely accounts for the high phosphate uptake by BS600 even in the slightly acidic environment (Fig. 5a). The increased point of zero charge further indicates an increase in the alkalinity (Lewis base) of the BS600 surface, which might induce additional hydrogen bonding and therefore be favorable for phosphate adsorption in a manner similar to that of phoslock adsorbent.22
 |
| | Fig. 6 Dependence of pHf on pHi during the equilibration of BS600 and B600. | |
3.3. Mechanism of phosphate adsorption to BS600
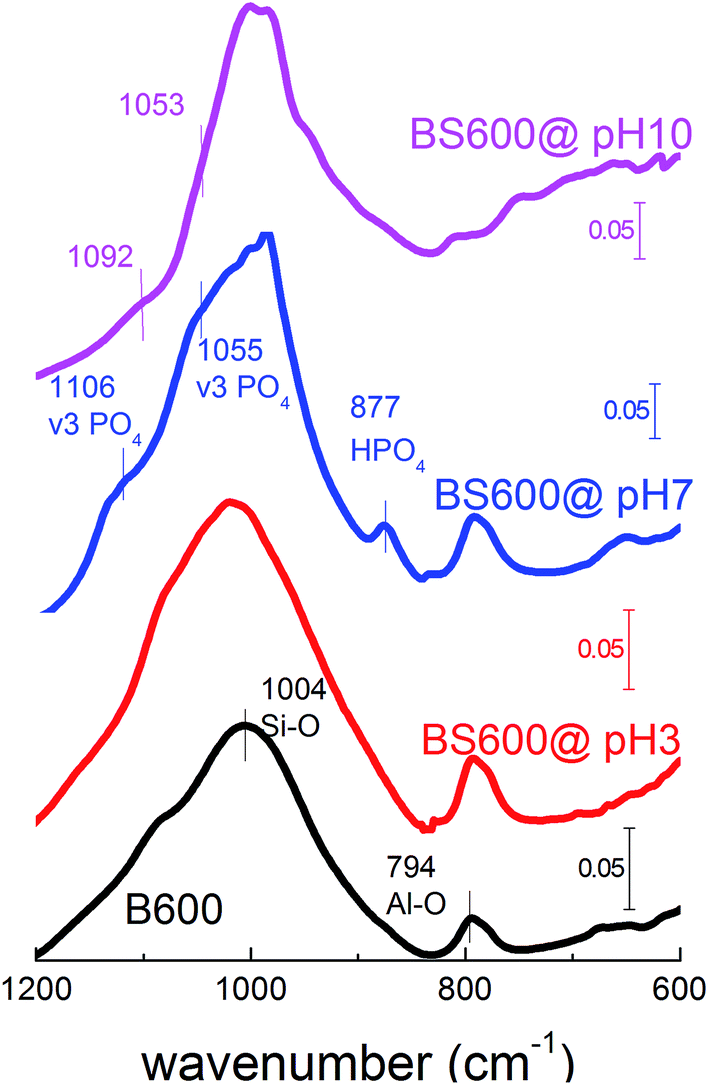
As mentioned above, sequestration of seawater Ca/Mg greatly increases the adsorption capacity and affinity of BS600 to environmental phosphate. Importantly, it is noted that the phosphate adsorption becomes less pH-sensitive, which is rather different from typical anion adsorption curves such as that of phosphate adsorption to Fe–Cu binary oxide,23 which is controlled by electrostatic interactions. As we mentioned above, the observed high affinity is likely to be due to the formation of a calcium phosphate configuration on the surface of BS600. As shown in Fig. 7, the EDX mapping clearly indicates that the hot zones of the adsorbed phosphate and the Ca-rich area are superimposed. This means that among Al, Mg and Ca, the latter is the determinant element controlling the uptake of phosphate, although AlPO4 is also identified as an important crystalline phase for phosphate adsorption.24 In the relevant study exploring the phosphate adsorption to Portland cement, it is suggested that the phosphate adsorption is mainly based on precipitation of the CaHPO4·2H2O crystalline phase.15 Unfortunately, none of the above-mentioned crystalline phases could be clearly identified by our additional XRD analyses (data not shown) because of their low surface content (qmax = 11.45 mg g−1). Alternatively, the wet clay samples (at the end of phosphate adsorption) were collected and characterized using the ATR-FTIR technique to identify in situ the configuration of the adsorbed phosphate (Fig. 8).
 |
| | Fig. 7 The SEM image and the corresponding element mapping of sample BS600. | |
 |
| | Fig. 8 FTIR spectra of B600 and BS600 collected at the end of phosphate adsorption at different pH values. | |
As shown in Fig. 8, the absorption bands at 1004 and 794 cm−1 appear in all samples, which originate from the Si–O and Al–O stretching model of aluminum phyllosilicate sheets of bentonite. For samples collected at the end of phosphate adsorption at pH 3 (BS600@pH 3), no significant absorption bands are noted other than those of aluminum phyllosilicate sheets of bentonite, which is probably due to the low phosphate adsorption at this pH environment (Fig. 5a). In contrast, the bands at 1106 and 1055 cm−1 appear in the FTIR spectrum of sample BS600@pH 7, which are assigned to the asymmetric stretching vibration (v3 mode) in PO4 groups.24 Along with the band centered at 877 cm−1, it is realized that the associated phosphate on the surface of BS600 is in the form of PO43− and HPO42−. At pH 10, the shoulders at 1053 and 1092 cm−1 suggest that PO43− is the major associated species on the surface of BS600 (BS600@pH 10). Importantly, although the appearance of these absorption bands clearly support that associated phosphate is in a configuration similar to that of the calcium phosphate phase, it does not directly indicate the formation of hydroxyapatite precipitates on the surface of modified bentonite. This is because other characteristic bands are likely to be overlapped within the Si–O stretching band, and it is difficult to extract them from the bentonite background owing to their low surface content. However, it is certain that the strong interaction between phosphate and surface calcium oxide allows BS600 to remove environmental phosphate in a much greener manner, which demonstrates the promising potential of our proposed modification method.
4. Conclusions
In this study, we demonstrated that the negatively charged bentonite surface can be effectively screened by the incorporation of seawater Ca/Mg, which allows the modified bentonite to remove negatively charged phosphate. In this manner, our results showed that modified bentonite exhibits a much higher adsorption capacity by two orders of magnitude compared to that of raw bentonite. It is thus believed that sequestrating seawater Ca/Mg would be a promising way to prepare green adsorbents for controlling the natural phosphate concentration in a large water body.
Acknowledgements
The authors are grateful for the support of this study by the Ministry of Science and Technology, ROC (MOST 104-2119-M-007-015 and MOST 104-2811-M-007-097) and Ningbo Nature Science Foundation (Grant No. 2015A610247 and 2015A610256).
References
- J. P. Gustafsson, L. B. Mwamila and K. Kergoat, Geoderma, 2012, 189–190, 304–311 CrossRef CAS.
- Y. L. Jan, T. H. Wang, M. H. Li, S. C. Tsai, Y. Y. Wei and S. P. Teng, Appl. Radiat. Isot., 2008, 66, 14–23 CrossRef CAS PubMed.
- S. Moharami and M. Jalali, Environ. Monit. Assess., 2014, 186, 6565–6576 CrossRef CAS PubMed.
- N. Yaghi and H. Hartikainen, Chemosphere, 2013, 93, 1879–1886 CrossRef CAS PubMed.
- K. Reitzel, F. O. Andersen, S. Egemose and H. S. Jensen, Water Res., 2013, 47, 2787–2796 CrossRef CAS PubMed.
- J. B. Zhou, P. X. Wu, Z. Dang, N. W. Zhu, P. Li, J. H. Wu and X. D. Wang, Chem. Eng. J., 2010, 162, 1035–1044 CrossRef CAS.
- S. M. Lee, Lalhmunsiama, Thanhmingliana and D. Tiwari, Chem. Eng. J., 2015, 270, 496–507 CrossRef CAS.
- T. H. Wang, C. J. Hsieh, S. M. Lin, D. C. Wu, M. H. Li and S. P. Teng, Environ. Sci. Technol., 2010, 44, 5142–5147 CrossRef CAS PubMed.
- M. J. Yang, J. W. Lin, Y. H. Zhan and H. H. Zhang, Ecol. Eng., 2014, 71, 223–233 CrossRef.
- J. Matusik, Chem. Eng. J., 2014, 246, 244–253 CrossRef CAS.
- M. Zamparas, A. Gianni, P. Stathi, Y. Deligiannakis and L. Zacharias, Appl. Clay Sci., 2012, 62–63, 101–106 CrossRef CAS.
- N. A. Oladoja, C. O. Aboluwoye, I. A. Ololade, O. L. Adebayo, S. E. Olaseni and R. O. A. Adelagun, Ind. Eng. Chem. Res., 2012, 51, 14637–14645 CrossRef CAS.
- Y. Yao, B. Gao, J. J. Chen, M. Zhang, M. Inyang, Y. C. Li, A. Alva and L. Y. Yang, Bioresour. Technol., 2013, 138, 8–13 CrossRef CAS PubMed.
- W. J. Liu, H. Jiang, K. Tian, T. W. Ding and H. Q. Yu, Environ. Sci. Technol., 2013, 47, 9397–9403 CrossRef CAS PubMed.
- X. J. Wang, J. D. Chen, Y. P. Kong and X. M. Shi, Water Res., 2014, 62, 88–96 CrossRef CAS PubMed.
- E. Ferrage, B. Lanson, B. S. Sakharov and V. A. Dtits, Am. Mineral., 2005, 90, 1358–1374 CrossRef CAS.
- S. Lazarevic, I. Jankovic-Castvan, D. Jovanovic, S. Milonjic, D. Janackovic and R. Petrovic, Appl. Clay Sci., 2007, 37, 47–57 CrossRef CAS.
- G. Rytwo, A. Banin and S. Nir, Clays Clay Miner., 1996, 44, 276–285 CrossRef CAS.
- T. H. Wang, M. H. Li, Y. Y. Wei and S. P. Teng, Appl. Radiat. Isot., 2010, 68(12), 2140–2146 CrossRef CAS PubMed.
- T. H. Wang, C. L. Chen, L. Y. Ou, Y. Y. Wei, F. L. Chang and S. P. Teng, J. Hazard. Mater., 2011, 192(3), 1079–1087 CrossRef CAS PubMed.
- T. H. Wang, T. Y. Liu, D. C. Wu, M. H. Li, J. R. Chen and S. P. Teng, J. Hazard. Mater., 2010, 173(1–3), 335–342 CrossRef CAS PubMed.
- L. Dithmer, A. S. Lipton, K. Reitzel, T. E. Warner, D. Lundberg and U. G. Nielsen, Environ. Sci. Technol., 2015, 49, 4559–4566 CrossRef CAS PubMed.
- G. Li, S. Gao, G. Zhang and X. Zhang, Chem. Eng. J., 2014, 235, 124–131 CrossRef CAS.
- C. M. Zhang, J. Yang, Z. W. Quan, P. P. Yang, C. X. Li, Z. Y. Hou and J. Lin, Cryst. Growth Des., 2009, 9, 2725–2733 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.