
Balancing intermediate state decay rates for efficient Pr3+ visible-to-UVC upconversion: the case of β-Y2Si2O7:Pr3+†
Ezra L. Cates*a and
Feifei Lib
aDepartment of Environmental Engineering and Earth Sciences, Clemson University, Clemson, SC, USA. E-mail: ecates@clemson.edu; Tel: +1-864-656-1540
bChemical and Biological Engineering, University of Colorado – Boulder, Boulder, CO, USA
First published on 22nd February 2016
Abstract
Materials that convert visible light into ultraviolet radiation have recently been studied for potential application to new catalytic and antimicrobial technologies. Conversion efficiencies of reported phosphors, however, have remained much lower than the more well-known IR-to-visible upconversion systems, due to a lack of lanthanide activator ions well suited for the required energy transfer mechanisms. Herein, the ceramic pyrosilicate β-Y2Si2O7:Pr3+ was prepared for the first time and its visible-to-UVC upconversion efficiency was optimized. The resulting material showed the most efficient upconversion of this type to date, with a 3.9-fold improvement over the compositionally similar X2-Y2SiO5:Pr3+ benchmark material. Analysis of both the steady-state and time-dependent luminescence behavior revealed that a relatively short-lived 3PJ intermediate state – normally considered detrimental to Pr3+ upconversion – in fact contributed to its efficacy. The pyrosilicate host also resulted in a much longer 1D2 luminescence lifetime, the origins of which are speculated to relate to the precise spacing of the 4f states. Based on the results, we discuss revised criteria for the “ideal” Pr3+ photophysical behavior and host requirements for maximizing UVC conversion efficiency.
Introduction
Upconversion (UC) materials include doped crystalline and organic molecular systems which achieve emission of one higher energy photon following the sequential absorption of two or more lower energy photons via intermediate excited states.1,2 The science surrounding infrared (IR)-to-visible UC phosphors has advanced rapidly since the birth of the NaYF4:Er3+,Yb3+ system in 1966,3 with this system now being explored for a variety of applications. These include bioimaging, drug delivery, and solar cells, among others.4–9 The ideal performance of Yb3+ as a sensitizer has been crucial to these developments, owing to its simple 4f electronic structure and ability to enhance absorption and energy transfer without inducing quenching of excited activator ions.1,2In visible-to-ultraviolet (UV) UC, however, no analogous lanthanide sensitizer exists and the field has struggled to demonstrate efficiencies suitable for applications that do not involve laser excitation. Such uses include enhancing spectral responses of UV-active photocatalysts,10 converting focused sunlight to germicidal UVC radiation (220–280 nm) for point-of-use water disinfection in the developing world,11,12 and converting ambient light to “trace” UVC emissions for light-activated antimicrobial surfaces.11,13 Successful proof-of-concept experiments having been published for most of these technologies; yet, unlike in IR-to-visible conversion, the materials' UC efficiencies have remained well below practical values. For applying this type of UC to biocidal applications, these challenges are somewhat offset by the high sensitivity of microorganisms to UVC photons, and the allure of technologies that could result from an ideal phosphor. The efficiency needed for a viable material is thus predicted to be on the order of 10−2%, compared to the 100% efficiency already achieved by existing IR-to-visible systems under low power excitation.12 Incremental advances in visible-to-UVC phosphor design may therefore prove to be a suitable route to valuable technologies in the future.
Activation with Pr3+ is the most effective option for achieving UVC emission using visible light excitation, through two-photon energy transfer UC (ETU) processes involving emission from the 4f5d band.14,15 Two mechanisms have been evidenced,16,17 and are shown in Fig. 1. An important criteria for Pr3+ visible-to-UVC systems is that the 4f5d band energy of the Pr3+ centers must be low enough for these mechanisms to occur; hence, hosts with low crystal field splitting that result in a high-energy 4f5d band position, such as fluorides, are not capable of such ETU.16 Historically, the [3PJ,3PJ] → [4f5d,3H4] mechanism (Fig. 1A, hereafter termed Mech. I) was assumed to be dominant, and phosphor development strategies were focused on increasing the 3PJ excited state lifetimes to enhance energy transfer between ions in these states.11,14,15 Such strategies include: (1) selection of host crystals with lower phonon cutoff energy to decrease 3P0 → 1D2 multiphonon relaxation rates and phonon-assisted [3P0,3H4] → [1D2,3H6] cross-relaxation;16 and (2) codoping with Li+ to disperse Pr3+ clusters, also alleviating cross-relaxation.18 The former approach was recently used by Cates et al.16 to yield the most efficient system to date, the oxyfluoride Lu7O6F9:Pr3+. Therein, the 3P0 lifetime was estimated at 15.5 μs, compared to 3.1 μs in the benchmark material X2-Y2SiO5:Pr3+,Li+ (YSO:Pr3+; both in ceramic form). Nonetheless, the gain in UC was lower than expected given the intermediate lifetime values, and we found that the [3PJ,1D2] → [4f5d,3H4] mechanism (Mech. II, Fig. 1B) was of greater significance in YSO:Pr3+ than previously assumed.16 While Mech. II involves the 1D2 state, which has a much slower decay rate, this was speculated to be offset by the poor spectral overlap of the respective ETU process.
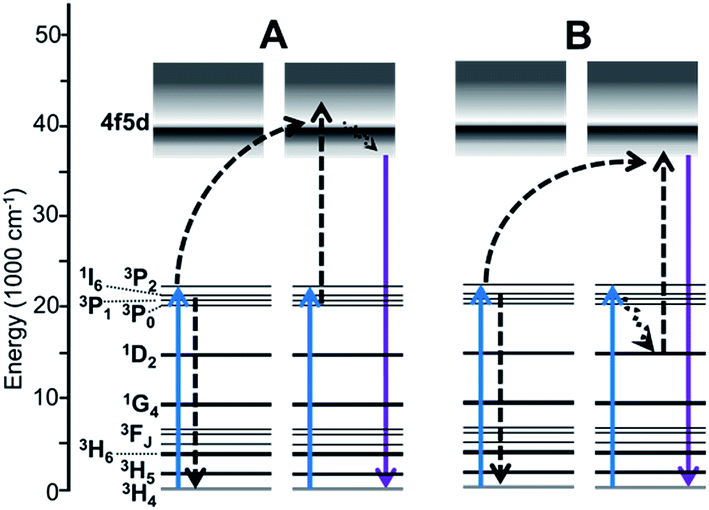 | ||
| Fig. 1 Established visible-to-UVC upconversion mechanisms of Pr3+-activated materials. (A) “Mech. I” involving two ions in the 3PJ intermediate states. (B) “Mech. II” involving a 3PJ → 1D2 relaxation event. Solid arrows depict light absorption or emission; dashed arrows depict energy transfer and non-radiative relaxation. | ||
Herein, we report on UC by the pyrosilicate β-Y2Si2O7:Pr3+ (YPS:Pr3+), which sets a new record for visible-to-UVC conversion efficiency. For the first time, we show evidence that Mech. II is the most efficient route to UVC anti-Stokes emission, even at the expense of Mech. I efficiency, and that 3PJ → 1D2 radiative or non-radiative relaxation can be beneficial.
Experimental section
Ceramics synthesis
YPS:Pr3+ ceramic samples were prepared in two stages, including sol gel synthesis of a precursor powder, followed by pressing the powder into pellets and sintering at higher temperature. The precursor powder preparation method described in ref. 13 for synthesis of YSO ceramics was employed, though in our case a higher molar fraction of tetraethylorthosilicate (TEOS, the Si source) was utilized to result in the pyrosilicate material. Pre-experiments found that a 60% stoichiometric excess of TEOS resulted in the best upconversion performance, possibly by stabilizing the β phase and discouraging other pyrosilicate polymorphs; thus a Y![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Si molar ratio of 1:1.6 was employed. For Pr3+ doping, a stoichiometric amount of Y3+ was omitted, while Li+ was treated as an additive (molar doping percentages herein are still expressed with respect to Y3+ stoichiometry). The precursor powder, fired at 600 °C in air, was then pressed into green bodies at 50 MPa using a 10 mm steel die and hydraulic pellet press. The amount of powder used per pellet was controlled at 0.28 g, which ultimately resulted in 1.2 mm-thick ceramics. Finally, the green bodies were sintered at 1300 °C in air for 3 h in alumina boats using a tube furnace. Undoped YPS ceramics were prepared using separate alumina boats to avoid cross-contamination of Pr3+ from doped samples. YSO:Pr3+(1.2%),Li+(10%) ceramics were prepared similarly according to published methods, also using a 10 mm die.13
:Si molar ratio of 1:1.6 was employed. For Pr3+ doping, a stoichiometric amount of Y3+ was omitted, while Li+ was treated as an additive (molar doping percentages herein are still expressed with respect to Y3+ stoichiometry). The precursor powder, fired at 600 °C in air, was then pressed into green bodies at 50 MPa using a 10 mm steel die and hydraulic pellet press. The amount of powder used per pellet was controlled at 0.28 g, which ultimately resulted in 1.2 mm-thick ceramics. Finally, the green bodies were sintered at 1300 °C in air for 3 h in alumina boats using a tube furnace. Undoped YPS ceramics were prepared using separate alumina boats to avoid cross-contamination of Pr3+ from doped samples. YSO:Pr3+(1.2%),Li+(10%) ceramics were prepared similarly according to published methods, also using a 10 mm die.13
Characterization
Powder X-ray diffraction (XRD) analysis was conducted with a Rigaku Miniflex using Cu K-α1/K-α2 emission. The XRD procedure was performed using the flat face of the as-prepared ceramics, without grinding. Rietveld analysis was conducted in order to estimate phase fractions, using GSAS and EXPGUI software.19,20 Scanning electron microscopy (SEM) images were obtained using a Hitachi S-4800 field emission microscope and gold sputter coating. Lithium content of the ceramics was analyzed by a commercial laboratory (NSL Analytical) using inductively couple plasma-mass spectrometry (ICP-MS) with sodium hydroxide digestion.Sample luminescence emission spectra were collected using a detection system identical one reported previously.16 Data covering large portions of the visible range were corrected using a photomultiplier response curve, obtained using a calibrated mercury reference lamp, since the response is not fully linear across this range. Blue light excitation was provided by a 1000 mW 447 nm diode laser (Opto Engine LLC), while UV and yellow-range excitation were provided by a tunable xenon lamp source (PowerArc, Horiba Scientific). The source housing and monochromator were purged with nitrogen to enhance <220 nm output. Unless otherwise indicated, laser-excited spectroscopic measurements were conducted using an appropriate band-pass filter to reduce the intrusion of laser light into the detector. Luminescence lifetimes were measured using an optical parametric oscillator coupled to a pulsed Nd:YAG laser (Continuum Surelite II), with pulse qualities and the detection system described in a previous work.16 Instrument response data were obtained using an undoped YPS ceramic to scatter the laser pulse and measure its decay profile in the absence of any luminescence. Multi-exponential decay equations of the form 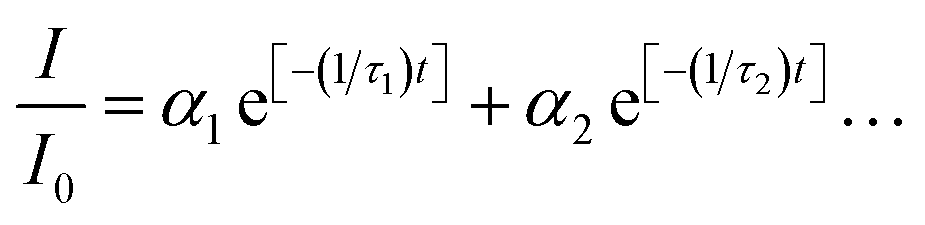 were fit iteratively in Excel, with fits shown in Fig. S1 and S2.† Therein, I/I0 is the fraction of emission intensity remaining, αn is the fractional contribution of the transition corresponding to that term, and τn is the respective lifetime value of that transition.
were fit iteratively in Excel, with fits shown in Fig. S1 and S2.† Therein, I/I0 is the fraction of emission intensity remaining, αn is the fractional contribution of the transition corresponding to that term, and τn is the respective lifetime value of that transition.
Results and discussion
The UC intensity of YPS:Pr3+ ceramics was maximized in preliminary experiments by assessing a range of activator and codopant concentrations, with YPS:Pr3+(1.5%),Li+(12%) ultimately giving the best results. Compared to samples prepared using the same method, but without Li+-codoping, 12 mol% Li+ resulted in a 74% enhancement in UC intensity (Fig. S3†). Increased Pr3+ UC efficiency resulting from Li+ codoping has been studied previously for YSO:Pr3+ powders, and was attributed to numerous effects on crystal structure and Pr3+ substitution;18 however, ICP-MS analysis herein showed that approximately 99% of added Li+ evaporated from the YPS:Pr3+ ceramics during the sintering process. Thus, the majority of UC enhancement observed may be attributed to a flux effect,21 and Li+ might be more accurately described as an additive in this case, rather than a dopant. A similar result of Li+ codoping was observed previously for preparation of YSO:Pr3+ ceramics, with ∼70% evaporating in that case.13Fig. 2A shows the results of XRD analysis for the optimized YPS:Pr3+ ceramic, with Rietveld refinement indicating largely phase-pure monoclinic β-Y2Si2O7 with no other pyrosilicate polymorphs detected. Cristobalite SiO2 was present at ∼5%, not suprisingly, due to an excess of Si added during synthesis. Fig. 2B displays SEM micrographs of the ceramic surface, which showed highly fused crystallites of up to ∼80 μm.
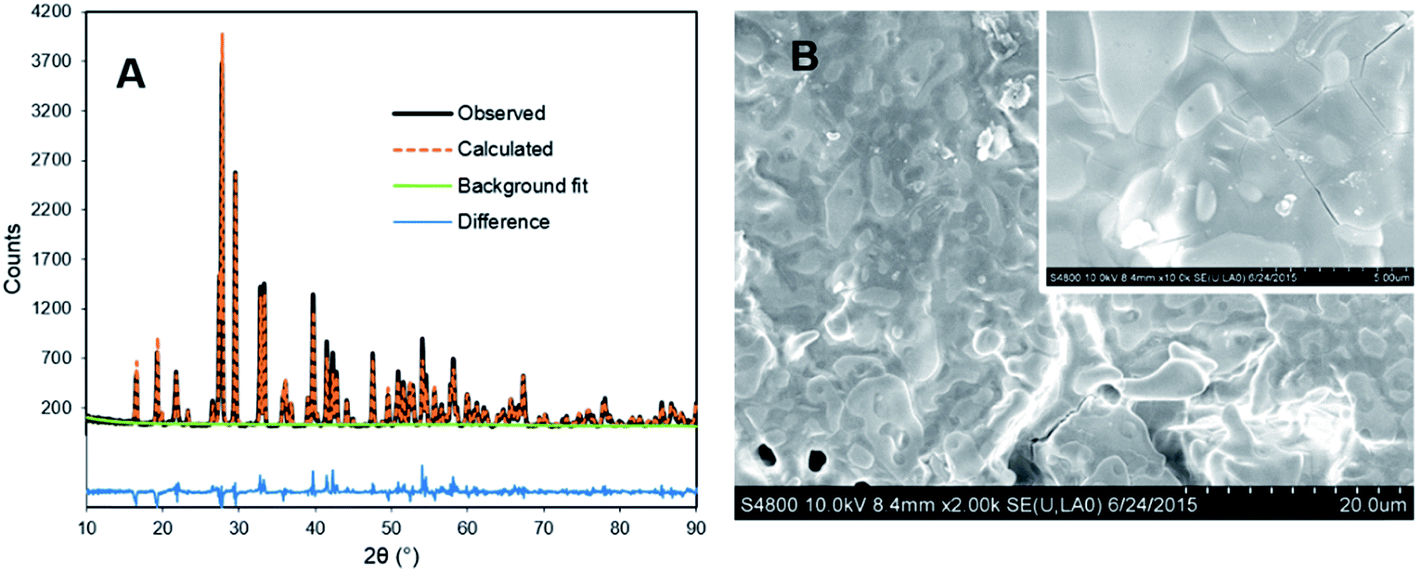 | ||
| Fig. 2 (A) XRD and Rietveld analysis results of YPS:Pr3+(1.5%),Li+(12%) ceramic prepared at 1300 °C. (B) SEM micrograph of the ceramic surface. Tick marks show 20 μm increments. Inset: higher magnification with 5 μm ticks. | ||
Fig. 3A shows the UC emission spectra under 447 nm laser excitation of the optimized YPS:Pr3+ ceramic and YSO:Pr3+ reference, of which the composition had also been optimized previously for UC efficiency.13 The YPS host resulted in a 3.1-fold increase in overall UC emission and 3.9-fold increase within the germicidal range (λ < 280 nm). Qualitatively, the YPS:Pr3+ primary emission peak was shifted toward higher energy and was slightly sharper than the YSO:Pr3+ data. These differences were ascribed to a lower degree of crystal field splitting in the less dense YPS22,23 and the presence of only one crystallographically distinct Y3+/Pr3+ site, respectively.22,24 The structure of YSO, on the other hand, offers two Y3+ sites23,24 and its wider Pr3+ emission spectrum thus comprises two similar, but overlapping, spectra. The UC excitation power dependence of YPS:Pr3+ (Fig. 3A inset) clearly indicated a two-photon UC process, with saturation occurring at higher laser powers.
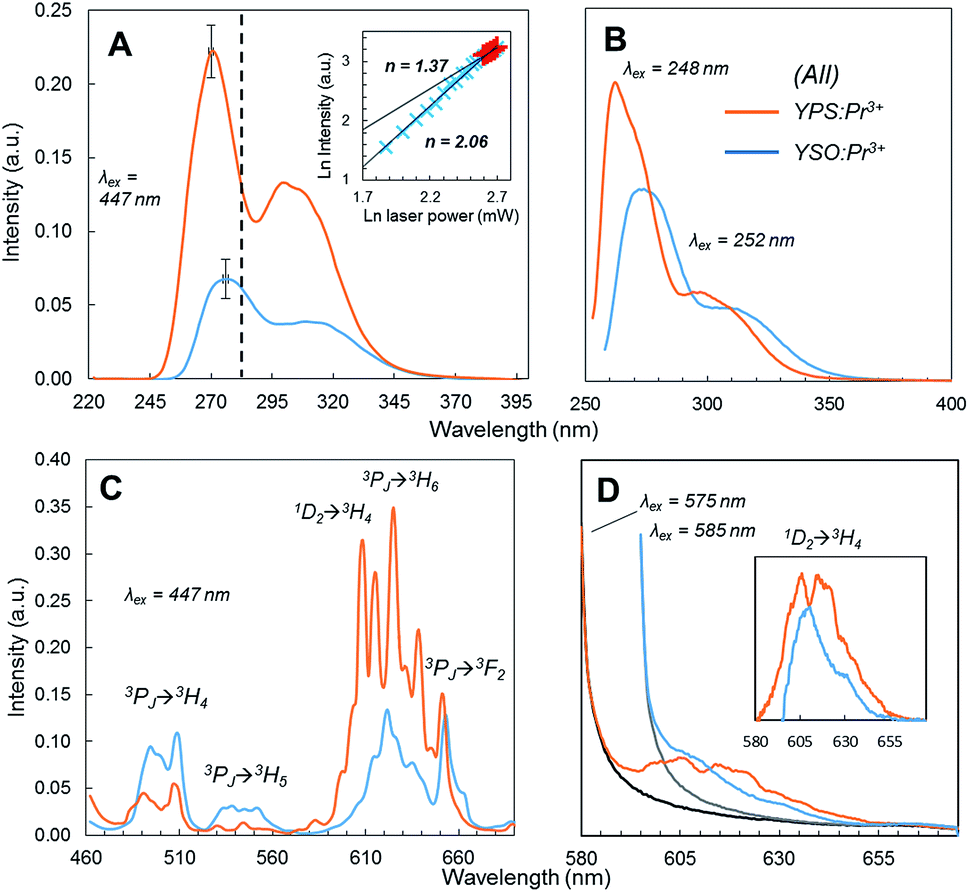 | ||
| Fig. 3 Emission spectra of optimized YPS:Pr3+ (orange) and YSO:Pr3+ (blue) ceramics. (A) UC emission under 447 nm excitation; dotted line shows upper boundary of UVC region; inset shows YPS:Pr3+ UC excitation power dependence data at high and low intensity regimes; slopes [n] of the linear regressions of the double-logarithmic data are indicated; error bars show the peak height standard deviation of three separately prepared samples. (B) Stokes emission under UV excitation. (C) Stokes emission under 3P2 (447 nm) excitation. (D) Stokes emission under 1D2 (575/585 nm) excitation; since no rejection filter was used, contribution from scattered excitation light was subtracted by measuring the spectra of undoped samples (black and gray series); inset shows the corrected 1D2 → 3H4 emission spectra. | ||
Fig. 3B shows the Pr3+ 4f5d emission of each ceramic under direct UV excitation by a xenon source at each material's respective excitation maximum. The YPS host still resulted in a higher emission intensity, but it was less drastic than the UC results and consistent with the higher optimized doping concentration of YPS:Pr3+ compared to the reference. Thus the much greater UC efficiency is apparently related to the ETU mechanism, and not due to differences in intrinsic 4f5d emission efficiency. Stokes emission from the 4f states induced by pumping the 3P2 level at 447 nm is shown in Fig. 3C. Compared to YSO:Pr3+, the yellow-red 1D2 → 4f emission was more dominant in the YPS:Pr3+ spectrum (note that 3PJ → 3H6/3F2 transitions also overlap with this wavelength range15). This difference in emitted spectral distribution could be explained by one of two possible causes: (1) significant quenching in one of the materials that reduced the emission intensity in a certain range; or (2) 3PJ → 1D2 relaxation occurred in YPS:Pr3+ to a much greater extent, with little difference in overall quenching between the two materials. To investigate, the 1D2 emission of YPS:Pr3+ and YSO:Pr3+ under direct excitation at 575 and 585 nm, respectively, was monitored and is shown in Fig. 3D. In contrast to the indirectly-excited 1D2 emission in Fig. 3C, these data showed much less of a difference in intensity, discounting significant preferential quenching. We therefore deduced that the different spectral distributions between the two ceramic systems seen under 447 nm excitation resulted mainly from 3PJ → 1D2 relaxation occurring to a greater extent in YPS:Pr3+. As described below, this finding has implications regarding the relative contributions of the two UC mechanisms, only one of which involves ions excited to the 1D2 level.
The results of luminescence decay measurements taken under pulsed laser excitation at various wavelengths are shown in Fig. 4 and associated lifetime values are displayed in Table 1. Decay fits may be seen in the (ESI, Fig. S1 and S2†). Upon exciting to 3P2 at 447 nm, YPS:Pr3+ emission at 490 nm showed single-exponential decay with a lifetime of 0.6 μs, attributed to the 3P0 → 3H4 transition. The YSO:Pr3+ ceramic, on the other hand, showed bi-exponential decay at 490 nm, assigned to the energetically equivalent 3P1 → 3H5 and 3P0 → 3H4 transitions and with much longer lifetimes of 1.9 and 3.7 μs, respectively. With such a short-lived intermediate state in YPS:Pr3+, the efficiency of Mech. I will undoubtedly be much lower for this system, and overall UC efficiency would thus be lower if this type of ETU were dominant.16 The UC results, however, show that this is not the case.
 | ||
| Fig. 4 Luminescence decay profiles of YPS:Pr3+ (black) and YSO:Pr3+ (gray) ceramics. (A) Decay of Stokes emission under 447 nm pulsed excitation. (B) Decay of Stokes emission under 580 nm pulsed excitation. (C) Decay of UC emission under 447 nm pulsed excitation; inset shows view with narrower y-axis and zoomed-out x-axis. | ||
| 3PJ lifetimes in μs | 1D2 lifetimes in μs | |
|---|---|---|
| YPS:Pr3+ | 0.60 (100%) | 1.3 (37%) |
| 9.1 (28.5%) | ||
| 38 (24%) | ||
| 83 (13.5%) | ||
| YSO:Pr3+ | 1.9 (78%) | 1.6 (60%) |
| 3.7 (22%) | 12 (34%) | |
| 56 (6%) |
The luminescence decay curves of the directly-excited 1D2 levels showed more complex behavior with triple- and quadruple-exponential decay (Fig. 4B, Table 1, Fig. S1C and S1D†). The lifetime of the 1D2 state is well known to be 1–2 orders of magnitude longer than other Pr3+ intra-4f transitions due to its spin-forbidden – in addition to parity-forbidden – nature.25,26 Yet our data in Table 1 show a large fraction of the emission at ∼610 nm for both ceramics following the 580 nm laser pulse was short-lived (100 μs) and more indicative of spin-allowed transitions. Since no spin-allowed transitions originating from 1D2 correspond to this energy, the source of these fast components is presently unclear. We theorize, however, that they arise from subpopulations of Pr3+ ions that were clustered in the material and considerably closer to one another than the rest of the activator ions. Non-radiative relaxation from 1D2 can occur via cross-relaxation proceeding as [1D2,3H4] → [1G4,3F3,4],27 and will be more efficient with less interionic separation. Once this subpopulation has undergone relaxation, the remaining more widely dispersed excited ions would continue to luminesce with slower decay. Regardless, it is clear from Fig. 4B that YPS:Pr3+ showed much slower overall 1D2 decay than YSO:Pr3+. The longest-lived component of its decay profile had a lifetime of 83 μs, compared to 56 μs for YSO:Pr3+ (we note that our values for YSO:Pr3+ lifetimes are more accurate than those we previously reported, which were estimated as singe-exponential16). The significantly slower 1D2 decay observed for YPS:Pr3+ is suggestive of a more prominent role of Mech. II in this phosphor.
Time dependent emission of UC luminescence by YPS:Pr3+ and YSO:Pr3+ is shown in Fig. 4C. The artifact peak apparent at ∼0.25 μs in both signals, as well as in the instrument response signal, was due to oscilloscope overshoot and is typical of high voltage measurements.28 Both materials showed similar decay of UC emission prior to 0.25 μs, after which YSO:Pr3+ showed slower decay lasting ∼3 μs. This observation is consistent with the 3P0 lifetime results which indicated a larger population of ions excited to the 3PJ level and available for Mech. I with YSO:Pr3+ than for YPS:Pr3+ during this time span. Both materials, however, had UC emission continuing well past 10 μs and the Fig. 4C inset shows that YPS:Pr3+ had the slowest such decay, continuing as long as 30 μs. While it is easy to misinterpret the greater integrated peak area of the YSO:Pr3+ signal in Fig. 4 to imply this system had more efficient UC, we stress that pulsed excitation produces different results than the continuous excitation conditions reflected in Fig. 3A. The data may be interpreted to suggest that under continuous pumping at much lower power, the 1D2 population and less-transient Mech. II in YPS:Pr3+ would accumulate to reach higher steady state values than in the YSO:Pr3+ system. The superior UC efficiency of YPS:Pr3+ can thus be attributed to a larger steady state 1D2 population due to: (1) faster rate of 3PJ → 1D2 decay; and (2) longer overall 1D2 lifetime. Of course, as seen in Fig. 1B, Mech. II still involves a Pr3+ ion excited to the 3PJ state; thus, there is a tradeoff between the longer 1D2 lifetime and the shorter 3PJ lifetime. Overall, the much longer 1D2 lifetime apparently is more than enough to compensate for the lower availability of 3PJ-excited ions during excitation of YPS:Pr3+.
The decay behavior of YPS:Pr3+ UC emission shown in Fig. 4C strongly indicates the occurrence of both mechanisms, evidenced by both quickly decaying and slowly decaying components. This association has previously been established for YSO:Pr3+.16 By analyzing these data we estimated lifetimes of 0.04 μs for the fast component of the YPS:Pr3+ signal and 0.2 μs for the slow component, which we attribute to mechanisms I and II, respectively. Compared to the former, UC by Mech. II requires a lower energy 4f5d band edge, such that the combined energies of two ions at 3PJ and 1D2 is sufficient to excite into the band during ETU (Fig. 1B). For example, UC in Lu7O6F9:Pr3+ was recently found to occur efficiently through Mech. I, while Mech. II was absent due to a high 4f5d band energy and lack of spectral overlap.16 Fig. 5 shows the 4f5d excitation spectrum of YPS:Pr3+, which provides a picture of the density distribution of 4f5d states and their energies, allowing further inspection of Mech. II in this material. Based on the data in Fig. 3D and C, respectively, we estimated the energy of the lowest 1D2 component and the 3P0 state in YPS:Pr3+ to be 16100 and 20200 cm−1. Following combination of these energies through ETU, the resultant total energy will be 36300 cm−1, corresponding to a wavelength of 275 nm. However, Fig. 5 indicates that this energy is below the band edge and no such ETU could occur. Therefore, Mech. II in YPS:Pr3+ must involve a more energetic 3PJ state than 3P0, such as 3P2. It is difficult to experimentally show the energy of the 3P2 state since its excitation and emission wavelengths are so closely spaced, though we probed its energy herein using pulsed excitation at 435 nm. This wavelength is expected to be only weakly absorbed via the 3H4 → 3P2 transition,16 however it gave less signal interference from the excitation pulse, as it is further from the detection wavelengths of interest. Decay signals were successfully resolved for detection wavelengths as low as 455 nm, shown in Fig. S2.† Combining the energy of this 3P2 Stark component and the lowest energy 1D2 state gives post-ETU energy of 38100 cm−1 (262 nm), which overlaps with the 4f5d excitation data and is thus a viable ETU mechanism.
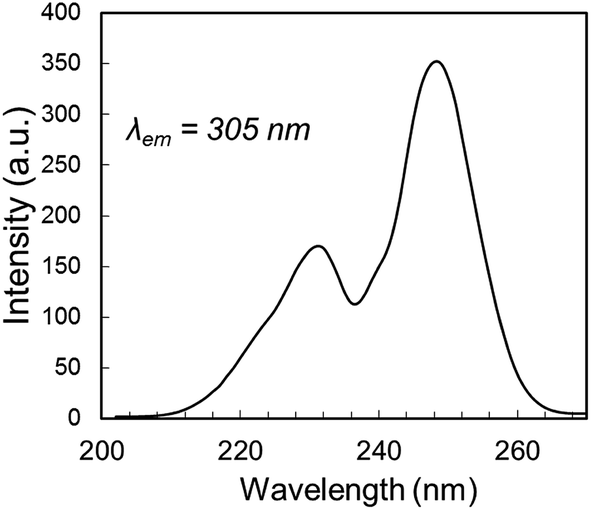 | ||
| Fig. 5 4f5d excitation spectrum of YPS:Pr3+, showing the 4f5d band edge energy at ∼37037 cm−1 (270 nm). | ||
As stated above, our results show that the high UC efficiency of YPS:Pr3+ is due to significant 3PJ → 1D2 relaxation, coupled with a long-lived 1D2 excited state; the causes of these phenomena themselves, however, have not yet been addressed. To explain the first phenomena, we note that the directly-excited 1D2 emission spectrum of YPS:Pr3+ in Fig. 3D covers a noticeably wider wavelength range than that of YSO:Pr3+. An energetically wider 1D2 manifold would provide greater spectral overlap for both variations of the 3PJ → 1D2 cross-relaxation mechanism, reducing the need for phonon assistance. Cross-relaxation efficiency is also heavily dependent on the interionic distance, and such effects should not be ruled out at this time. The reason for slower 1D2 luminescence decay in YPS:Pr3+ than in YSO:Pr3+ may be similarly related to the specific relative energies of their respective 4f Stark states, as non-radiative relaxation from 1D2 can also occur via cross-relaxation proceeding as [1D2,3H4] → [1G4,3F3,4].27
In our previous work, which compared YSO:Pr3+ to Lu7O6F9:Pr3+, we noted that Pr3+ visible-to-UVC conversion can be improved by selecting a host with minimal phonon cutoff energy while maintaining a low-energy Pr3+ 4f5d band edge.16 This combination was expected to minimize non-radiative losses while encouraging proper spectral overlap for Mech. II. Interestingly, YPS:Pr3+ has both higher phonon cutoff energy than YSO:Pr3+ (1100 cm−1 vs. 970 cm−1)29,30 and a higher-energy 4f5d band edge, defying both of these criteria. Efficient ETU was instead achieved in this system through the combination of fast 3PJ → 1D2 relaxation and long-lived 1D2 excited states, yielding the most efficient UVC-range Pr3+ UC known to date. Given the reported external conversion efficiency of 0.0019% for YSO:Pr3+ ceramics under direct fluorescent lamp excitation,13 we expect YPS:Pr3+ to show an efficiency several times greater under similar excitation conditions. This gain represents a significant advancement in this class of UC materials, though additional improvements are required for practical application to biocidal or catalytic technologies using low power excitation. The results of this study support selection of future host materials that result in rapid 3PJ → 1D2 decay, while still conforming to the other criteria that have been uncovered over the last 5 years. With the growing list of design tools for improving Pr3+ UC efficiency, phosphor systems suitable for practical use may indeed be achievable.
Conflict of interest
The authors declare no competing financial interests.Acknowledgements
We wish to thank Dr Jaehong Kim of Yale University for use of the pulse laser system.References
- F. Auzel, Upconversion and Anti-Stokes Processes with f and d Ions in Solids, Chem. Rev., 2004, 104, 139–174 CrossRef CAS PubMed.
- F. Wang and X. Liu, Recent Advances in the Chemistry of Lanthanide-Doped Upconversion Nanocrystals, Chem. Soc. Rev., 2009, 38, 976–989 RSC.
- F. Auzel, Comptes Rendus deomptes Rendus de l'Académie des Sciences B Physics, 1966, p. 263 Search PubMed.
- S. Fischer, J. C. Goldschmidt, P. Loper, G. H. Bauer, R. Bruggemann, K. Kramer, D. Biner, M. Hermle and S. W. Glunz, Enhancement of Silicon Solar Cell Efficiency by Upconversion: Optical and Electrical Characterization, J. Appl. Phys., 2010, 108, 044912 CrossRef.
- T. Trupke, M. A. Green and P. Wurfel, Improving Solar Cell Efficiencies by Up-Conversion of Sub-band-gap Light, J. Appl. Phys., 2002, 92(7), 4117–4122 CrossRef CAS.
- F. Li, C. Li, X. Liu, Y. Chen, T. Bai, L. Wang, Z. Shi and S. Feng, Hydrophilic, Upconverting, Multicolor, Lanthanide-Doped NaGdF4 Nanocrystals as Potential Multifunctional Bioprobes, Chem.–Eur. J., 2012, 18, 11641–11646 CrossRef CAS PubMed.
- F. Li, C. Li, J. Liu, X. Liu, L. Zhao, T. Bai, Q. Yuan, X. Kong, Y. Han, Z. Shi and S. Feng, Aqueous Phase Synthesis of Upconversion Nanocrystals Through Layer-by-Layer Epitaxial Growth for In Vivo X-Ray Computed Tomography, Nanoscale, 2013, 5, 6950–6959 RSC.
- P. Zhang, W. Steelant, M. Kumar and M. Scholfield, Versatile Photosensitizers for Photodynamic Therapy at Infrared Excitation, J. Am. Chem. Soc., 2007, 129, 4526–4527 CrossRef CAS PubMed.
- N. M. Idris, M. K. Gnanasammandhan, J. Zhang, P. C. Ho, R. Mahendran and Y. Zhang, In Vivo Photodynamic Therapy Using Upconversion Nanoparticles as Remote-Controlled Nanotransducers, Nat. Med., 2012, 18, 1580–1585 CrossRef CAS PubMed.
- E. L. Cates, S. L. Chinnapongse, J.-H. Kim and J.-H. Kim, Engineering Light: Advances in Wavelength Conversion Materials for Energy and Environmental Technologies, Environ. Sci. Technol., 2012, 46, 12316–12328 CrossRef CAS PubMed.
- E. L. Cates, M. Cho and J.-H. Kim, Converting Visible Light into UVC: Microbial Inactivation by Pr3+-Activated Upconversion Materials, Environ. Sci. Technol., 2011, 45, 3680–3686 CrossRef CAS PubMed.
- E. L. Cates and J.-H. Kim, Bench-scale evaluation of water disinfection by visible-to-UVC upconversion under high-intensity irradiation, J. Photochem. Photobiol., B, 2015, 153, 405–411 CrossRef CAS PubMed.
- S. L. Cates, E. L. Cates, M. Cho and J.-H. Kim, Synthesis and Characterization of Visible-to-UVC Upconversion Antimicrobial Ceramics, Environ. Sci. Technol., 2014, 48, 2290–2297 CAS.
- C. Hu, C. Sun, J. Li, Z. Li, H. Zhang and Z. Jiang, Visible-to-ultraviolet upconversion in Pr3+:Y2SiO5 crystals, Chem. Phys., 2006, 325, 563–566 CrossRef CAS.
- C. L. Sun, J. F. Li, C. H. Hu, H. M. Jiang and Z. K. Jiang, Ultraviolet Upconversion in Pr3+:Y2SiO5 Crystal by Ar+ Laser (488 nm) Excitation, Eur. Phys. J. D, 2006, 39, 303–306 CrossRef CAS.
- E. L. Cates, A. P. Wilkinson and J.-H. Kim, Visible-to-UVC Upconversion Efficiency and Mechanisms of Lu7O6F9:Pr3+ and Y2SiO5:Pr3+ Ceramics, J. Lumin., 2015, 160, 202–209 CrossRef CAS.
- E. L. Cates and J.-H. Kim, Upconversion Under Polychromatic Excitation: Y2SiO5:Pr3+,Li+ Converts Violet, Cyan, Green, and Yellow Light into UVC, Opt. Mater., 2013, 35, 2347–2351 CrossRef CAS.
- E. L. Cates, A. P. Wilkinson and J.-H. Kim, Delineating Mechanisms of Upconversion Enhancement by Li+ Codoping in Y2SiO5:Pr3+, J. Phys. Chem. C, 2012, 116, 12772–12778 CrossRef CAS.
- B. H. Toby, EXPGUI. A Graphical User Interface for GSAS, J. Appl. Crystallogr., 2001, 34, 210–213 CrossRef CAS.
- A. C. Larson and R. B. Von Dreele, General Structure Analysis System (GSAS); LAUR, 1994, pp. 86–748 Search PubMed.
- Z. Sun, Y. Zhou and M. Li, Effect of LiYO2 on the Synthesis and Pressureless Sintering of Y2SiO5, J. Mater. Res., 2008, 23, 732–736 CrossRef CAS.
- G. J. Redhammer and G. Roth, β-Y2Si2O7, a New Thortveitite-Type Compound, Determined at 100 and 280 K, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2003, 59, i103–i106 CrossRef.
- B. A. Maksimov, V. V. Ilyukhin, Y. A. Khariton and N. V. Belov, Crystal Structure of Yttrium Oxyorthosilicate, Y2O3·SiO2 = Y2SiO5: Dual Function of Yttrium, Crystallogr. Rep., 1971, 15, 806 Search PubMed.
- E. Dell'Orto, M. Fasoli, G. Ren and A. Vedda, Defect-Driven Radioluminescence Sensitization in Scintillators: The Case of Lu2Si2O7:Pr, J. Phys. Chem. C, 2013, 117, 20201–20208 CrossRef.
- S. Mahlik, M. Malinowski and M. Grinberg, High Pressure Luminescence and Time Resolved Spectra of La2Be2O5:Pr3+, Opt. Mater., 2011, 34, 164–168 CrossRef CAS.
- K. D. Oskam, A. J. Houtepen and A. Meijerink, Site Selective 4f5d Spectroscopy of CaF2:Pr3+, J. Lumin., 2002, 97, 107–114 CrossRef CAS.
- R. Naccache, F. Vetrone, A. Speghini, M. Bettinelli and J. A. Capobianco, Cross-Relaxation and Upconversion Processes in Pr3+ Singly Doped and Pr3+/Yb3+ Codoped Nanocrystalline Gd3Ga5O12: The Sensitizer/Activator Relationship, J. Phys. Chem. C, 2008, 112, 7750–7756 CrossRef CAS.
- D. S. Anker and L. D. Merkle, Ion–ion Upconversion Excitation of the 4f5d Configuration in Pr:Y3Al5O12: Experiments and Förster Theory-based Rate Equation Model, J. Appl. Phys., 1999, 86, 2933–2940 CrossRef CAS.
- J. Sokolnicki, Upconversion Luminescence from Er3+ in Nanocrystalline Y2Si2O7:Er3+ and Y2Si2O7:Yb3+,Er3+ Phosphors, Mater. Chem. Phys., 2011, 131, 306–312 CrossRef CAS.
- A. A. Kaminskii, S. N. Bagayev, K. Ueda, J. Dong and H. J. Eichler, New Passively Q-switched LD-pumped Self-Raman Laser with Single-step Cascade SE → SRS Wavelength Conversion on the Base of Monoclinic Nd3+:Y2SiO5 Crystal, Laser Phys. Lett., 2010, 7, 270–279 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra01121g |
| This journal is © The Royal Society of Chemistry 2016 |