DOI:
10.1039/C6RA01087C
(Paper)
RSC Adv., 2016,
6, 28015-28022
Selective oxidation of organic sulfides to sulfoxides using sugar derived cis-dioxo molybdenum(VI) complexes: kinetic and mechanistic studies†
Received
13th January 2016
, Accepted 3rd March 2016
First published on 17th March 2016
Abstract
Six Mo(VI) complexes of 4,6-O-ethylidene-β-D-glucopyranosylamine derived ligands have been used for the selective oxidation of five organic sulfides to corresponding sulfoxides. The structure of a new sulfide [(((2-(phenylthio)phenyl)imino)methyl)naphthalen-2-ol] (S5) and its corresponding sulfoxide (SO5) has been established using single crystal X-ray diffraction studies along with other routine analytical techniques. The yields of sulfoxides were calculated using HPLC and one of the catalysts was recycled five times without any appreciable loss in its activity. Kinetic studies on sulfoxide formation was performed on compound S5 using UV-visible spectroscopy, which suggested overall first order kinetics. A plausible mechanism for sulfide oxidation has been proposed based on our findings and previous literature reports.
1. Introduction
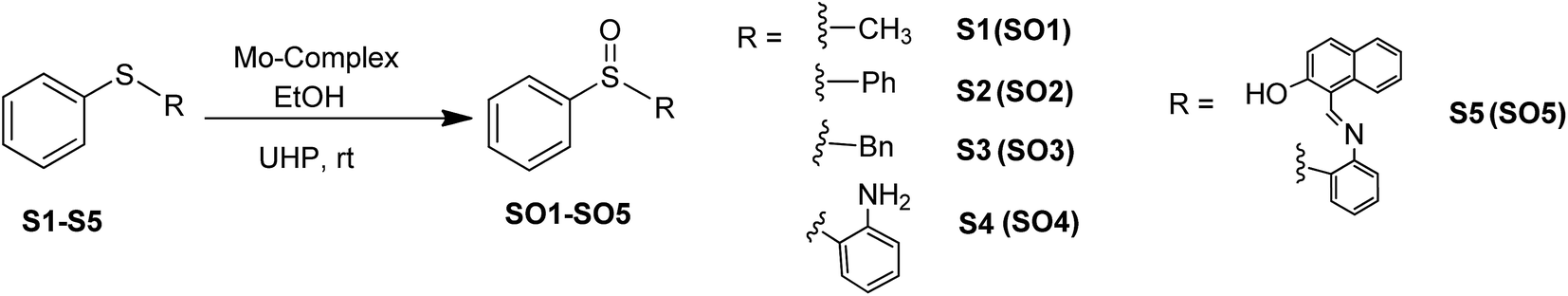
Sulfoxides are important from a biological as well as a chemical point of view due to their utility in drug development,1 asymmetric synthesis2 and oxo-transfer reagents like that in Swern oxidation.3 They are used in the preparation of several biologically and pharmaceutically important compounds like compactin, ML-236A (antihyperlipidimic agent),4 tacrolimus or FK-506 (immunosuppressive agent),5 11-oxoequilenin methyl ether (steroid),6 and 12,13-epoxytrichothec-9-ene (having a wide range of biological properties)7 etc. In the context of organic synthesis, sulfoxides are used in carbon–carbon bond formation,8–10 the Diels–Alder reaction11–13 etc. Due to the versatile use of sulfoxides, their preparation from corresponding organic sulfides are well documented, however, the methods have several drawbacks like over-oxidation of the substrate to sulfone, longer reaction times, selectivity problems, and the use of strong oxidising agents like hydrogen peroxide (H2O2) and meta-chloroperoxybenzoic acid (m-CPBA)14–17 etc. Hence, the need of the current scenario is to develop an environmentally benign protocol having sustainable and greener reaction conditions. In this venture, we are developing the methodology for selective oxidation of organic sulfides into corresponding sulfoxides using glucose derived cis-dioxo Mo(VI) complexes as the catalyst. At the initial stage, we had optimized the conditions for the oxidation of thioanisole (S1) to methyl phenyl sulfoxide (SO1) with respect to the solvent, catalytic loading, reaction timing etc., and the findings have been already communicated.18 The best yield of sulfoxide was isolated from the reaction performed in ethanol, using commercially available urea–hydrogen peroxide (UHP) as a mild oxidant.
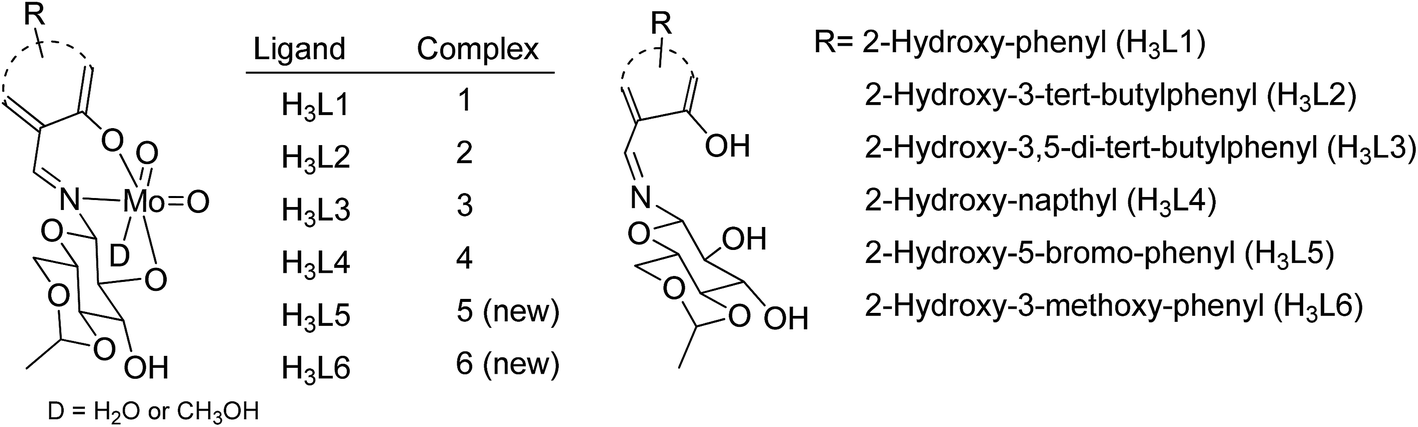
Molybdenum is one of the essential and less toxic metals, which easily form complexes with a variety of biologically important compounds like carbohydrates,18,19 porphyrins,20,21 flavins22,23 and amino acids.24 It plays a vital role in the activities of xanthine oxidase, nitrate reductase, nitrogenase, DMSO reductase25,26 etc. Our initial success in the selective oxidation of S1 to SO1 using the glucose derived cis-dioxo molybdenum(VI) complex under mild conditions, prompted us to make the methodology sustainable and reproducible for general organic sulfides. Along this line, we have used six complexes (Fig. 1), including two new ones as a catalyst for selective oxidation of organic sulfides to corresponding sulfoxides (Scheme 1). Five different sulfides were tested for oxidation, and the (%) formation of sulfoxides were obtained using the HPLC method. Turn over number and turn over frequency (per hour) were also calculated for all the reactions and the same were found in the range of 7.4–19.6 and 12–78 respectively.
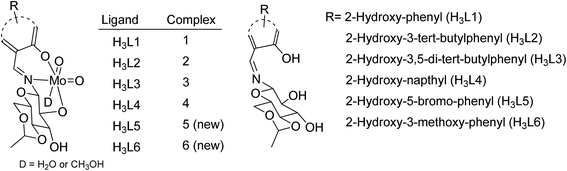 |
| | Fig. 1 cis-Dioxo molybdenum(VI) complexes of H3Ln. | |
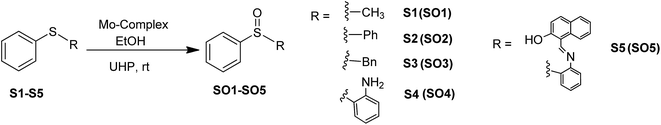 |
| | Scheme 1 Synthetic protocol of sulfoxides using sugar derived Mo(VI) complexes. | |
Kinetic studies were performed on the oxidation of S5 using UV-visible spectroscopy and several control reactions were performed to ascertain the reaction paths of sulfide oxidation. Based on our current studies and the reported literature, we have proposed a plausible mechanism for this reaction. Hence, this paper deals with the synthesis of a new sulfide, selective catalytic oxidation of organic sulfides to sulfoxides using sugar derived cis-dioxo Mo(VI) complexes and mechanistic aspects of this oxidation reaction.
2. Experimental
2.1. General
5-Bromosalicylaldehyde, ortho-vanillin, 2-(phenylthio)aniline (S4), 2-hydroxy-1-napthaldehyde and UHP were procured from Sigma-Aldrich India, while diphenyl sulfide (S2), benzyl phenyl sulfide (S3) and molybdenum(VI) oxide bis(2,4-pentanedionate) [MoO2(acac)2] were from Alfa Aeser. The complexes 5 and 6 were synthesized following the procedure reported for complex 1.27 All other chemicals and solvents were purchased from local vendors and solvents were purified and dried following the standard methods before use. All the experiments were performed at room temperature under normal atmospheric conditions.
UV-visible absorption spectra were recorded on a Shimadzu UV-260 spectrophotometer and FTIR spectra on an ABB Bomen MB 3000 FTIR machine using a KBr Matrix. 1H and 13C NMR spectra were recorded in DMSO-d6 and CDCl3 on a Bruker Avance spectrometer and data was processed using MestReNova software. HRMS and ESI-MS were recorded on a Thermo scientific Q EXACTIVE and Hewlett-Packard’ HP GS/MS 5890/5972 mass spectrometer respectively. The yields of sulfoxides were calculated by HPLC separation using a Waters Sunfire C-18 column of 5 μm, 4.6 × 250 mm, a flow of 0.85 mL min−1; wavelength of 315 nm; temperature of 28 °C; injection volume of 20 μL; eluent of water/MeOH (v/v) 0.18/0.67. Calibration was done using 30, 40, 50, 60, 70 and 80 ppm, of sulfoxide solutions.
Single crystal X-ray diffraction data were collected on a Rigaku XtaLAB mini diffractometer using graphite monochromated Mo-Kα radiation. An empirical absorption correction was applied on the data and all calculations were performed using the CrystalStructure28 crystallographic software package except for refinement, which was performed using SHELX-97.29 The crystallographic figures were generated using ORTEP 3v2 (ref. 30) and mercury 3.3.31
2.2. Preparation of complex 5
To a stirring solution of H3L5 (0.1176 g, 0.30 mmol) in methanol (5 mL), MoO2(acac)2 (0.0978 g, 0.30 mmol) was added at room temperature and the reaction was continued for 13 h. The workup was done similarly to that reported for complex 2,18 to isolate the yellow solid product. Yield: 0.134 g (81%); mp: chared at 208–210 °C; UV-vis [λmax; nm (ε; L cm−1 mol−1) in DMSO]: 260 (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 511), 348 (1980); IR (KBr; cm−1): 3371, 1643, 1088, 903. 1H NMR (DMSO-d6, 400 MHz, ppm): δ 8.57 (1H, s, HC
511), 348 (1980); IR (KBr; cm−1): 3371, 1643, 1088, 903. 1H NMR (DMSO-d6, 400 MHz, ppm): δ 8.57 (1H, s, HC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N), 8.04 (1H, br, ArH), 7.61 (1H, d, J = 6.0 Hz, ArH), 6.89 (1H, d, J = 7.6 Hz, ArH), 5.65 (1H, br, glucose OH), 4.76 (2H, m, glucose H-1, ethylidene CH), 4.13 (4H, m, glucose H-5, methanolic-CH3), 3.80–3.05 (5H, m, glucose, H-2, H-3, H-4, H-6a,b), 1.26 (3H, br, ethylidene CH3); 13C NMR (DMSo-d6, 100 MHz, ppm): δ 161.4 (CN), 158.7, 137.7, 136.8, 123.1, 122.1, 110.7, 99.3, 91.2, 85.6, 81.0, 73.4, 69.9, 67.6, 49.0, 20.6; HRMS: m/z calcd for (M + H)+ C15H17BrMoNO8 515.9192; found 515.9358, without solvent.
N), 8.04 (1H, br, ArH), 7.61 (1H, d, J = 6.0 Hz, ArH), 6.89 (1H, d, J = 7.6 Hz, ArH), 5.65 (1H, br, glucose OH), 4.76 (2H, m, glucose H-1, ethylidene CH), 4.13 (4H, m, glucose H-5, methanolic-CH3), 3.80–3.05 (5H, m, glucose, H-2, H-3, H-4, H-6a,b), 1.26 (3H, br, ethylidene CH3); 13C NMR (DMSo-d6, 100 MHz, ppm): δ 161.4 (CN), 158.7, 137.7, 136.8, 123.1, 122.1, 110.7, 99.3, 91.2, 85.6, 81.0, 73.4, 69.9, 67.6, 49.0, 20.6; HRMS: m/z calcd for (M + H)+ C15H17BrMoNO8 515.9192; found 515.9358, without solvent.
2.3. Preparation of complex 6
This compound was prepared following the procedure adopted for complex 5, but using (H3L6) (0.1020 g, 0.30 mmol) and MoO2(acac)2 (0.0978 g, 0.30 mmol). Yield: 0.130 g (89%); orange solid; mp: chared 233–235 °C; UV-vis [λmax; nm (ε; L cm−1 mol−1) in DMSO]: 287 (12336), 361 (2347); IR (KBr; cm−1): 3333, 1643, 1088, 903. 1H NMR (DMSO-d6, 400 MHz, ppm): δ 8.52 (1H, s, HCN), 7.32 (1H, d, J = 7.2 Hz, ArH), 7.19 (1H, d, J = 7.6, ArH), 6.91 (1H, m, ArH), 5.63 (1H, s, glucose OH), 4.72 (2H, m, ethylidene CH, glucose H-1), 4.18 (1H, m, glucose H-5), 3.90–3.10 (8H, m, glucose H-2, H-3, H-4, H-6a,b, methanolic-CH3), 1.26 (3H, br, ethylidene CH3); 13C NMR (DMSO-d6, 100 MHz, ppm): δ 159.39 (CN), 152.3, 149.5, 126.0, 121.3, 119.8, 117.2, 99.3, 91.1, 85.4, 81.1, 73.5, 69.9, 67.7, 56.2, 20.7; HRMS: m/z calcd for (M + H)+ C16H19MoNO9 467.0114; found 467.0102, without solvent.
2.4. Synthesis of [(((2-(phenylthio)phenyl)imino)methyl)naphthalen-2-ol] (S5)
To the ethanolic solution of 2-hydroxynapthaldehyde (1.01 mmol), S4 (1 mmol) was added and the reaction mixture was refluxed for 8 h. The reaction mixture was cooled and the resultant yellow crystalline solid product was filtered, washed with diethyl ether and dried under vaccum. Yield: 86%, yellow crystalline solid; mp: 140–142 °C; IR (KBr; cm−1) UV-vis [λmax; nm (ε; L cm−1 mol−1) in ethanol]: 315 (5599), 442 (4817), 465 (4531). 1H NMR (CDCl3, 400 MHz, ppm): δ 15.24 (1H, d, J = 2.4 Hz, ArOH), 9.40 (1H, d, J = 2.4 Hz, HCN), 8.13 (1H, d, J = 8.4 Hz, ArH), 7.84 (1H, d, J = 9.2 Hz, ArH), 7.76 (1H, d, J = 7.6 Hz, ArH), 7.54 (1H, m, ArH), 7.48–7.14 (11H, m, ArH); 13C NMR (CDCl3, 100 MHz, ppm) δ 167.6, 155.8, 145.7, 136.2, 134.0, 133.1, 132.2, 131.5, 131.1, 129.3, 129.3, 128.1, 128.0, 127.6, 127.5, 127.0, 123.6, 121.4, 119.1, 118.1, 109.3. Slow evaporation of the hexane solution of S5 yielded X-ray suitable single crystals and the structure was established using single crystal X-ray crystallography.
2.5. General procedure for selective oxidation of organic sulfides
To a stirring suspension of molybdenum complexes (1–6; 0.05 mmol) and sulfides (1 mmol) in 3 mL of ethanol, UHP (1 mmol) was added at room temperature. The progress of the reaction was monitored by thin layer chromatography and upon completion of the reaction, yields were calculated by HPLC (Fig. S7–S9, ESI†).
2.6. Synthesis of [(((2-(phenylsulfinyl)phenyl)imino)methyl)naphthalen-2-ol] (SO5)
This compound was prepared following the general procedure described above for sulfide oxidation, using S5 (1 mmol), UHP (1 mmol) and complex 1 (0.05 mmol). After stirring the reaction mixture for 40 minutes at room temperature, pure SO5 was separated from the reaction mixture using column chromatography on a silica gel solid support and ethyl acetate/hexane (30/70 v/v) as the eluent. Yield: 46%, yellow crystalline solid; mp: 142–143 °C; UV-vis [λmax; nm (ε; L cm−1 mol−1) in ethanol]: 331 (5699), 386 (7598). 1H NMR (CDCl3, 400 MHz, ppm) δ 14.37 (1H, s, ArOH), 9.33 (1H, s, HCN), 8.19 (1H, dd, J = 7.6, 1.7 Hz, ArH), 8.09 (1H, d, J = 8.4 Hz, ArH), 7.95 (1H, d, J = 9.2 Hz, ArH), 7.83 (1H, d, J = 8.0 Hz, ArH), 7.68 (2H, m, ArH), 7.63–7.54 (3H, m, ArH), 7.43 (1H, m, ArH), 7.35–7.25 (5H, m, ArH); 13C NMR (CDCl3, 100 MHz, ppm) δ 163.6, 159.9, 145.8, 145.0, 139.1, 136.3, 132.7, 132.2, 131.1, 129.4, 129.2, 128.2, 127.9, 127.6, 125.7, 124.6, 123.9, 119.8, 119.3, 118.6, 109.4. Slow diffusion of hexane into a dichloromethane solution of SO5 afforded X-ray suitable single crystals and finally the structure was confirmed by single crystal X-ray crystallography.
2.7. Catalyst recycling
Recycling of the catalyst was performed using complex 1 (0.05 mmol), S4 (1 mmol) and UHP (1 mmol) in ethanol (3 mL) at room temperature. At the end of the first cycle of the reaction, the solvent was evaporated under reduced pressure and the pasty mass was extracted with dichloromethane (3 × 3 mL) to remove the sulfide and its oxidized products. The residue (complex 1) was freshly treated with an equimolar amount of sulfide and UHP as mentioned above for the next cycle. The organic portion of the extract was used for isolation of the product using column chromatography.
2.8. Kinetic studies
A main reaction of complex 1 (0.005 mmol), sulfide S5 (0.11 mmol), and UHP (0.11 mmol) in ethanol 10 (mL) was set at room temperature. 20 μL of the reaction mixture was diluted to a final volume of 5 mL and its absorbance was measured at 465 nm using a UV-visible spectrophotometer. A decrease in the concentration of S5 during the reaction at 298 K was monitored using a pre-generated calibration curve and the data was used to establish the order of the reaction.
3. Results and discussion
3.1. Synthesis and characterization of two new complexes
H3L5 and H3L6 were reacted with MoO2(acac)2 in methanol to yield the corresponding sugar containing molybdenum(VI) complexes. The formation of the complexes were confirmed by FTIR, UV-visible, NMR and Mass spectroscopy. FTIR studies of both complexes exhibited strong characteristic vibration bands at 903, 1088, 1643 and 3333–3371 cm−1 corresponding to νMoO, νC–O, νCN and νO–H respectively. No appreciable changes were observed for νC–O and νCN stretching frequencies of complexes with respect to their corresponding free ligands, but a substantial change was observed in the νO–H region of the spectra. Sharp bands in the OH region of the ligands converted into a broad one after the metalation reaction, indicating the increment in hydrogen bonding interactions. The peak at 13.01 and 5.33 ppm corresponding to the phenolic and C2-hydroxyl proton of glucose respectively in the ligand, disappeared from the 1H NMR spectra of the molybdenum complexes revealing the metal complexation via the phenolate and alcohalate group. In both complexes, the signal of glucose C3–OH shifted down field with respect to the free ligands, which might be attributed to the change in electronic environment contributed by the chelated metal ion. All these spectral changes are consistent with the spectral description of reported similar Mo(VI) complexes of H3Ln (n = 1–4).18,27
3.2. Selective oxidation of sulfide to sulfoxide
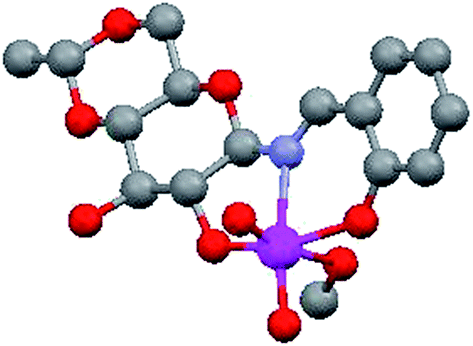
The molecular structure of 4,6-O-ethylidene-N-(2-hydroxybenzylidene)-β-D-glucopyranosylamine (H3L1) derived Mo(VI) complex (Fig. 2) has already been established27 and a similar complex has also been reported by Zhao et al.,32 using a glucosamine derived ligand. The literature is rich with the catalytic reactions of di-oxo Mo(VI) complexes,33–35 however it is scarce for sugar derived from such molecules. Zhao et al. published catalytic epoxidation reactions,32 we have reported the selective synthesis of a series of bis(indolyl)methanes by condensing indole derivatives with carbonyl compounds,36 while Mohammadnezhad et al.37 and our group have recently communicated the oxidation of S1 to SO1.18 We had reported the optimization of reaction conditions with respect to catalytic loading, solvent effect, reaction time etc. on a single substrate S1. In continuation of our previous work, we have tried to develop a general protocol for sulfide oxidation by sugar derived cis-dioxo Mo(VI) complexes and, in the process, the number of catalysts as well as substrates have been increased. All the reactions were performed under optimized conditions developed by us for sulfoxide formation,18 i.e., ethanol as the reaction solvent and a shorter reaction time (15 min) using a 1:1 molar ratio of sulfides and UHP. In order to better understand the conversion and purity, we have calculated the yields of product formation using HPLC and details of the method is stated in the experimental section. We have achieved the conversion with good to excellent yields and the same is summarized in Table 1. The formation of SO1–SO4 was established by comparing the respective melting points, IR, NMR and ESI-MS (characterization data and Fig. S10–S18: ESI†) spectra with the reported literature.17,18,38,39 Sheikhshoaie et al. have also done similar reactions using a 2-[(2-hydroxypropylimino)methyl]phenol derived Mo(VI) complex as the catalyst and UHP as a mild oxidant40 and our results are comparable to them. Varma and Naicker have reported the oxidation of S1 using UHP without any catalyst but at 85 °C and the yield was 80%.41 So, the use of a catalyst has not only reduced the reaction temperature but also improved the yield and purity of the product as they noticed a 10% mixture of sulfone.
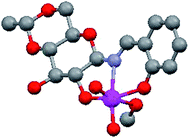 |
| | Fig. 2 Structure of 4,6-O-ethylidene-N-(2-hydroxybenzylidene)-β-D-glucopyranosylamine (H3L1) derived cis-dioxo Mo(VI) complex.27 | |
Table 1 Summary of sulfide oxidation using D-glucose derived cis-dioxo molybdenum(VI) complexes as the catalysta and UHP as the mild oxidizing agent
| Entry |
Complex |
Sulfoxide |
Yieldb (%) |
TON |
TOF/h |
| Refer to Fig. 1 for the structure of catalysts. Yield (%) was calculated by HPLC after 15 min of reaction time. Yield (%) was calculated by HPLC after 40 min of reaction time. |
| 1 |
1 |
SO2 |
90 |
17.9 |
72 |
| 2 |
|
SO3 |
90 |
18.1 |
72 |
| 3 |
|
SO4 |
98 |
19.6 |
78 |
| 4 |
|
SO5 |
50 |
10 |
15 |
| 5 |
2 |
SO2 |
79 |
15.8 |
66 |
| 6 |
|
SO3 |
89 |
17.8 |
72 |
| 7 |
|
SO4 |
96 |
19.2 |
78 |
| 8 |
|
SO5 |
37c |
7.4 |
12 |
| 9 |
3 |
SO2 |
86 |
17.1 |
66 |
| 10 |
|
SO3 |
89 |
17.7 |
72 |
| 11 |
|
SO4 |
94 |
18.7 |
72 |
| 12 |
|
SO5 |
44c |
8.9 |
12 |
| 13 |
4 |
SO2 |
89 |
17.7 |
72 |
| 14 |
|
SO3 |
93 |
18.5 |
72 |
| 15 |
|
SO4 |
98 |
19.6 |
78 |
| 16 |
|
SO5 |
45c |
8.9 |
12 |
| 17 |
5 |
SO2 |
75 |
15 |
60 |
| 18 |
|
SO3 |
82 |
16.4 |
66 |
| 19 |
|
SO4 |
91 |
18.1 |
72 |
| 20 |
|
SO1 |
90 |
18 |
72 |
| 21 |
|
SO5 |
61c |
12.2 |
18 |
| 22 |
6 |
SO2 |
82 |
16.5 |
66 |
| 23 |
|
SO3 |
83 |
16.7 |
66 |
| 24 |
|
SO4 |
84 |
17 |
66 |
| 25 |
|
SO1 |
88 |
17.5 |
72 |
| 26 |
|
SO5 |
66c |
13.2 |
18 |
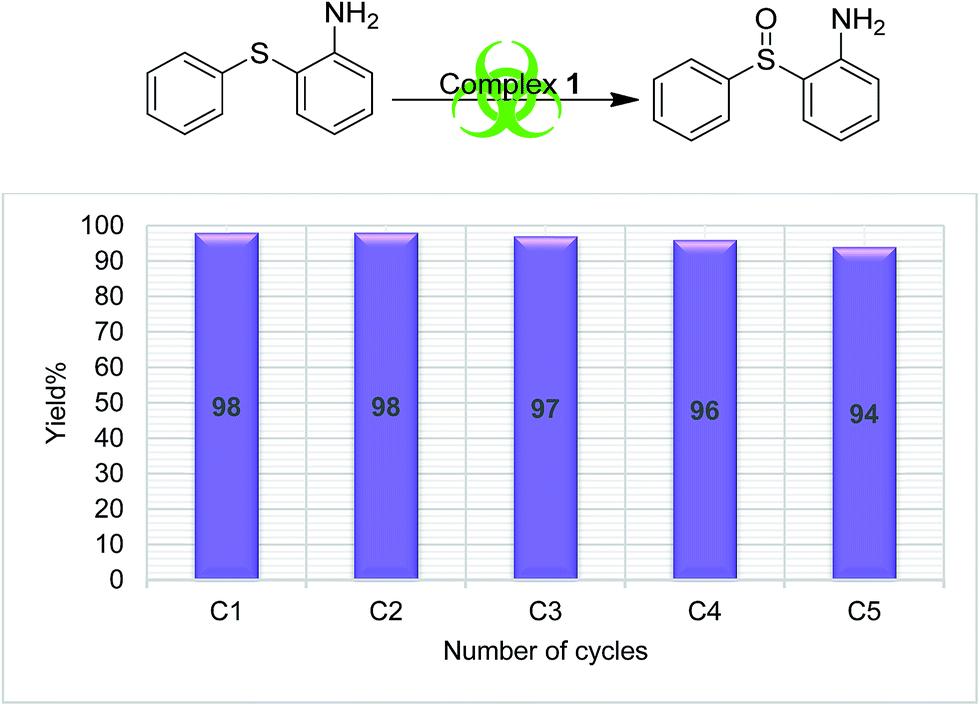
In order to understand the stability of catalysts during the catalytic reaction, a recyclability test was performed using complex 1 and S4 as a substrate. The catalyst was recycled five times (methods explained in Experimental section), and the results are presented in Fig. 3, which clearly supports the reliability of the catalytic process. No appreciable change in the catalytic efficiency was observed as the sulfoxide formation was in the range of 98 to 94%, from the first cycle to the end of the fifth cycle respectively.
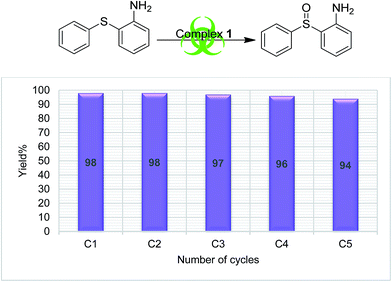 |
| | Fig. 3 Graphical representation of catalytic recyclability using complex 1 on S4. | |
The literature supports that it is easy to oxidize organic sulfides having electron-donating substituents than one having electron withdrawing groups.42 Our finding also followed this trend as we found the lowest yields (37–66%) for the oxidation of S5, while the highest (84–98%) were found for compound S4. The salient feature of the present sulfoxidation protocol is its high selectivity to form only sulfoxide and neither sulfones nor an oxidized benzylic product with substrate S3.
3.3. Kinetic studies
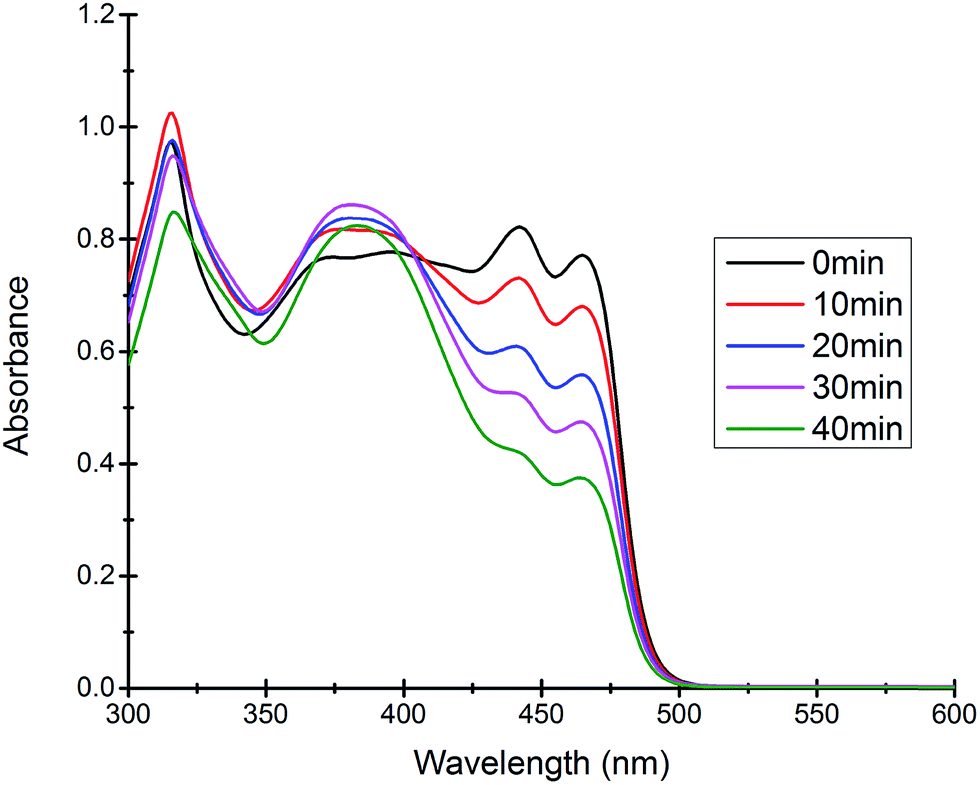
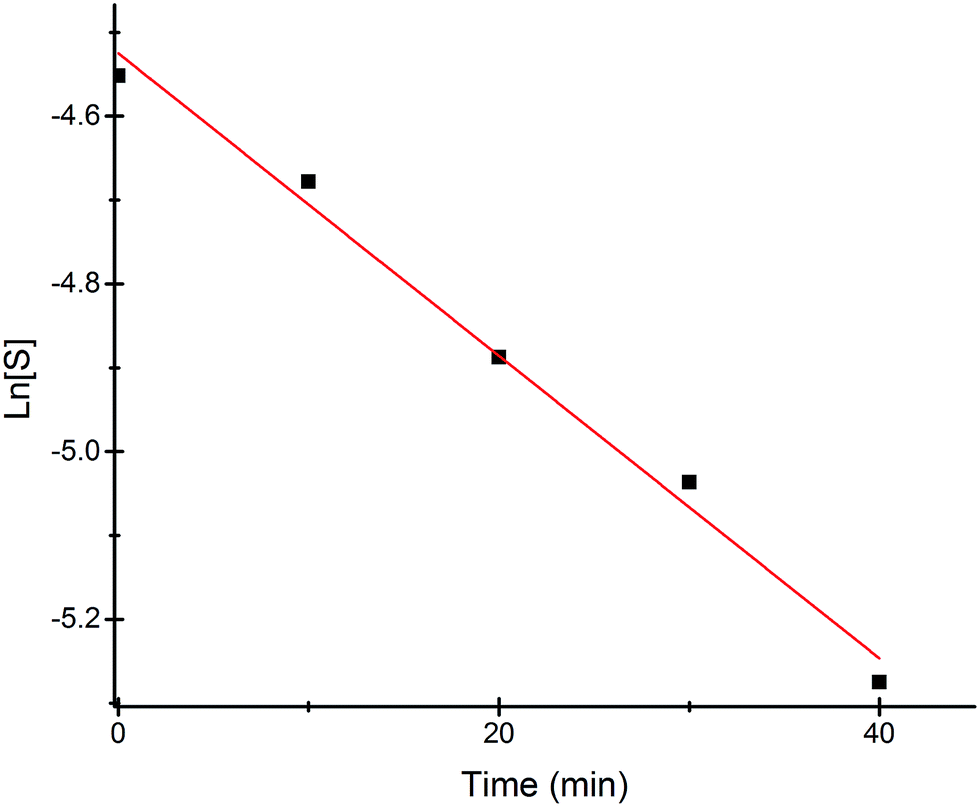
Most of the commercially available general organic sulfides and their corresponding sulfoxides exhibit a common overlapping absorption band in the UV-visible spectra. In our case, the spectral pattern of S1–S4 behaved similarly and the rate of oxidation was also fast. So, in order to understand the overall order of the reaction, we planned to develop a sulfide which should have a slow rate of oxidation along with a non-overlapping absorption band in the UV-visible spectrum for the substrate and corresponding oxidized product. In this venture, we synthesized a new sulfide S5, which fulfilled both criteria and hence the kinetics of the reaction was explored on this molecule using UV-visible spectroscopy. A comparison of UV-visible absorption spectra of the reactants (Fig. S-23, ESI†) revealed that only S5 absorbs at 465 nm and hence the oxidation reaction was monitored by measuring the absorbance of this band with course of time at 298 K. A decrease in absorbance at 465 nm was measured for the first 40 min (Fig. 4) and the data was analyzed. A linear ln[S5] vs. time plot (Fig. 5) supported the overall first order reaction kinetics with a rate constant, k = 0.01804 min−1 and a half-life, t1/2 = 38.4 minutes.
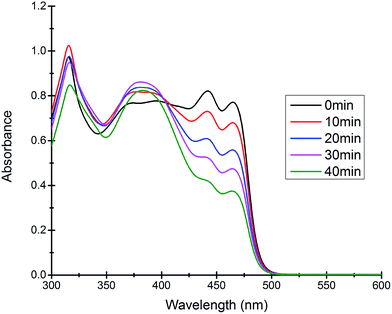 |
| | Fig. 4 Time dependent UV-visible spectra of the reaction mixture revealing the gradual decrease in the absorption band at 465 nm due to the consumption of the substrate S5. | |
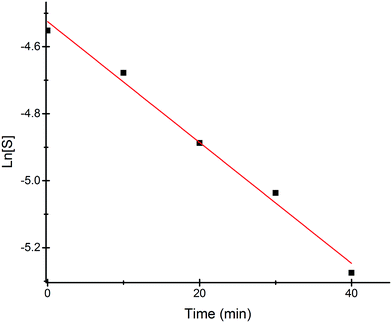 |
| | Fig. 5 Linear fitting plot of ln[S5] vs. time at 298 K. | |
3.4. Mechanism of reaction
In order to gain insight into the reaction mechanism, sulfide S2 was added to the ethanolic solution of the molybdenum complex 1 while stirring. No visual change in the physical appearance of the reaction mixture was noticed even after 15 min of stirring. The reaction mixture was analyzed using a UV-visible spectrophotometer (Fig. S-24, ESI†), however no interaction between complex 1 and sulfide S2 was established. Similar studies performed using an ethanolic solution of sulfide S2 and UHP (Fig. S-25, ESI†) also failed to establish the interactions. Finally, molybdenum complex 1 was suspended in ethanol and UHP was added to that, which resulted in an instantaneous yellowish transparent solution (Fig. 6). The UV-visible absorption studies (Fig. S-26, ESI†) on these sets of control reactions also supported the interaction via the hypochromic effect along with an overall change in the peak positions.
 |
| | Fig. 6 (A) Complex 1 in ethanol and (B) complex 1 in ethanol after UHP addition. | |
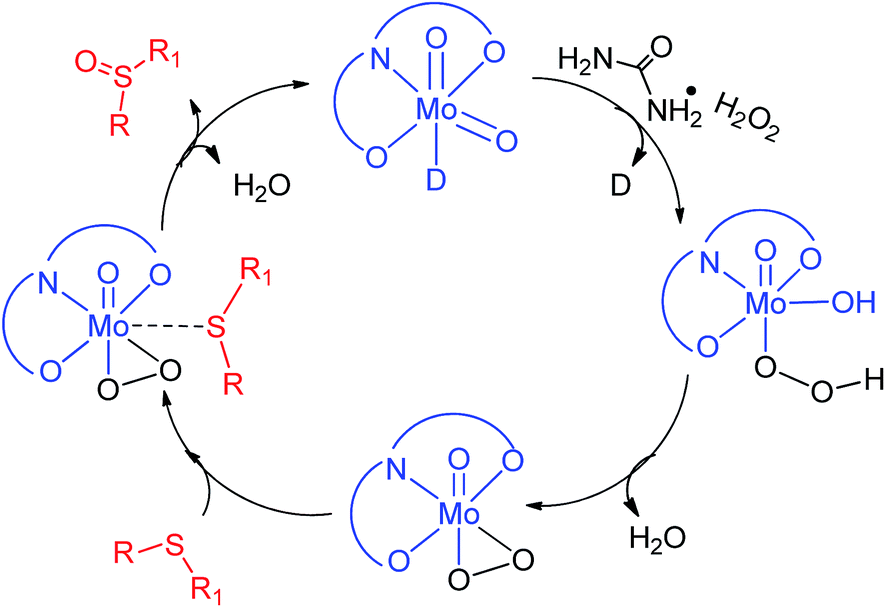
Furthermore, the effect of the variation in UHP concentration was studied, where no change in the kinetics of the reaction was noticed, however an increase in the conversion of sulfides into sulfones was observed. All these studies concluded that the binding of UHP with the molybdenum complex occurs at the initial stage of the sulfide oxidation reaction. A similar finding has also been reported by Kamata et al.,43 Panda et al.44 and Chakravarthy et al.45 using W, Ti and Mo complexes respectively, however all of them have used H2O2 as an oxidizing agent. Chakravarthy et al. have proposed the possibility of sulfide binding with either a molybdenum center or peroxo oxygen.45 The literature reveals that an increase in electron density about sulfur due to its attachment with electron donor groups increases the oxidation efficiency and we also noticed the same trend. This fact is only possible if sulfide is interacting with a metal center and not with peroxo oxygen, as the electronegative nature of oxygen will repel the electron rich sulfur centre. Hence, in our opinion, sulfide ion binds with the Mo center rather than peroxo oxygen. On the basis of our results and the previous literature reports43–45 a plausible oxidation mechanism has been summarized in Scheme 2.
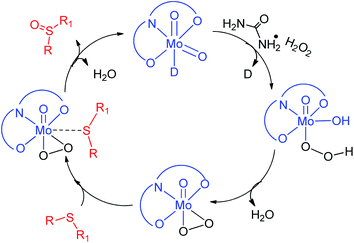 |
| | Scheme 2 Proposed mechanistic pathway for the formation of sulfoxide. | |
3.5. Crystal structure of S5 and SO5 and their correlation with the oxidation process
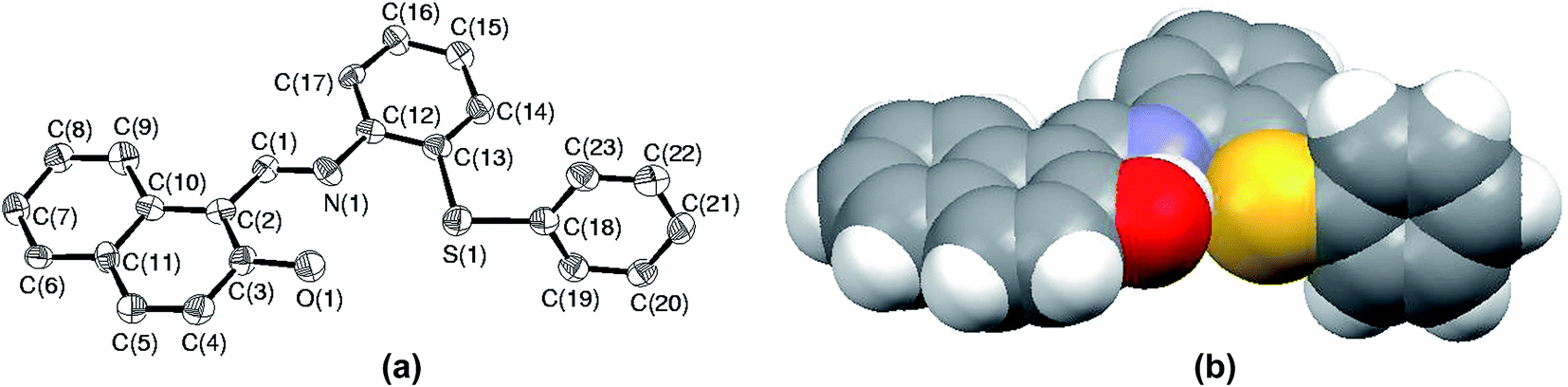
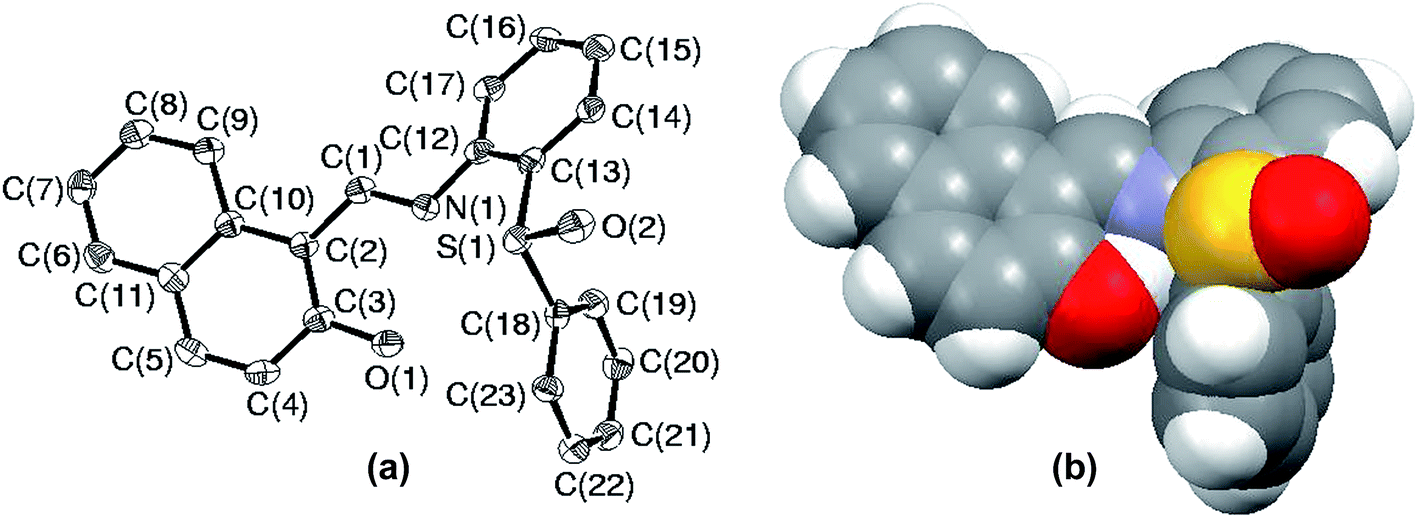
Among all the sulfides used in this paper, the rate of oxidation as well as yields were the lowest for S5. The low yield is attributed to the formation of sulfoxide at the initial stage followed by parallel formation of sulfide and sulfones, while the low rate of oxidation might be due to either steric crowding or the electronic environment or both together about the sulfur center. In order to understand this chemistry, we crystallized both the sulfide (S5) and corresponding sulfoxide (SO5) and their solid state structure was established using single crystal X-ray crystallography. In both cases, crystal quality was averaged and the R values (0.0910 and 0.0627) were within the accepted range for publication as mentioned in the crystallographic summary (Table 2). The ORTEP plot of S5 shown in Fig. 7(a) indicates the hindrance of the sulfide ion to come closer to the Mo center of the catalyst and the same can be clearly viewed in the space-filling model (Fig. 7(b)). The whole molecule is arranged in space such that three sides of the basal plane of sulfur is covered with naphthyl and phenyl rings, while the perpendicular arrangement of one of the phenyl rings obstructs its exposure to the catalyst from the vertical side. Such an atomic environment about sulfur might be responsible for its slow oxidation process as it might not be able to interact with the catalytic center appropriately.
Table 2 Summary of the crystallographic data and structural parameters of S5 and SO5
| |
S5 |
SO5 |
| Empirical formula |
C23H17NOS |
C23H17NO2S |
| Molecular weight |
355.43 |
371.43 |
| T (K) |
100 |
100 |
| Crystal system |
Orthorhombic |
Orthorhombic |
| Space group |
P212121 |
P212121 |
|
| Cell constants |
| a (Å) |
6.132(2) |
5.6260(10) |
| b (Å) |
14.536(5) |
12.886(2) |
| c (Å) |
19.327(7) |
24.399(5) |
| V (Å3) |
1722.6(11) |
1768.8(6) |
| Z value |
4 |
4 |
| Dcalcd (g cm−3) |
1.371 |
1.395 |
| Total reflections |
11726 |
15463 |
| Unique reflections |
3922 |
4029 |
| Parameters |
236 |
245 |
| Final R (I > 2σ(I)) |
0.0910 |
0.0627 |
| Rw |
0.2135 |
0.1231 |
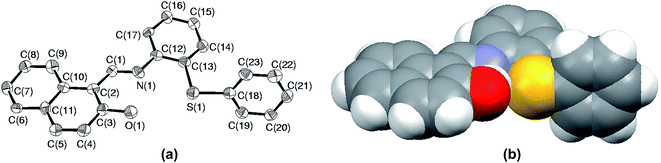 |
| | Fig. 7 (a) ORTEP plot of S5 drawn using 50% probability ellipsoids; (b) space-filling model of S5 showing the hindrance about the sulfur atom. | |
Since the environment about sulfur is crowded in S5, it raises a curiosity to know about the special arrangement of molecular fragments in SO5. In order to fulfill this inquisitiveness, the solid state structure of SO5 was explored using single crystal X-ray crystallography. The ORTEP plot of SO5 with an atomic labeling scheme is presented in Fig. 8(a), while Fig. 8(b) illustrates its space-filling model. This structure clearly reveals the overall change in the atomic environment about sulfur, which facilitates it to protrude out, which might help it in converting to sulfone parallel to sulfide oxidation. The changes in conformations of sulfide and sulfoxide can be easily established by analyzing the selected torsion angles. The angles C(1)N(1)C(12)C(17), C(10)C(2)C(1)N(1), C(14)C(13)S(1)C(18) and C(13)S(1)C(18)C(19) are 15.9(11), 179.4(7), 9.5(8) and −113.5(7) in sulfide, while −36.0(7), 170.6(4), −106.0(4) and 6.6(5) respectively in sulfoxide.
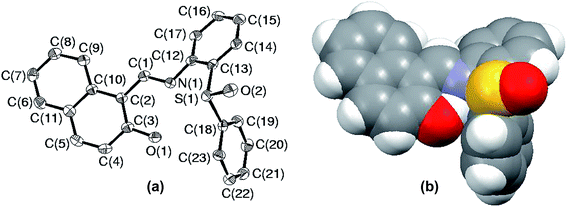 |
| | Fig. 8 (a) ORTEP plot of SO5 drawn using 50% probability ellipsoids; (b) space-filling model of compound SO5. | |
4. Conclusions
Selective oxidation of organic sulfides to sulfoxides have been explored using glucose derived cis-dioxo molybdenum(VI) complexes as catalysts and urea–hydrogen peroxide as a mild oxidising agent. A total of six catalysts were tested on five sulfides and good to excellent conversion was achieved under milder reaction conditions and shorter reaction time. The conversion was best for compound S4 (84–98%) and least for compound S5 (37–66%). All the yields were calculated using HPLC separation and the purity of the products was further checked by comparing their melting point, FTIR and NMR data with the literature values. A new sulfide S5 and corresponding sulfoxide SO5 also has been developed and their structures have been established using the single crystal X-ray diffraction method. One of the catalysts was tested for recyclability and only a minute change (∼4% loss) in activity was observed during five cycles, revealing the sustainability of the catalyst under the reaction conditions. Even though similar reactions are reported using other catalysts and conditions, the advantage of our report is the greener methodology i.e., catalyst consisting of bio-viable species (D-glucose and molybdenum), short reaction time, high selectivity, room temperature reaction and ethanol as the reaction medium. This paper also deals with the kinetics and mechanistic aspects of sulfide oxidation.
Acknowledgements
We are thankful to Dr Angshuman Roy Choudhury for his help in crystallographic data collection. A. K. Sah is grateful to Department of Science and Technology (DST) and University Grants Commission (UGC) for the financial support. The Department of Chemical Sciences, Indian Institute of Science Education and Research, Mohali is acknowledged for providing the XtaLABmini X-ray diffractometer facility. We are also thankful to DST FIST and UGC SAP for their financial support in procuring the instruments and developing the research infrastructure.
References
- M. C. Carreño, Chem. Rev., 1995, 95, 1717–1760 CrossRef.
- I. Fernández and N. Khiar, Chem. Rev., 2003, 103, 3651–3705 CrossRef PubMed.
- A. M. Khenkin and R. Neumann, J. Am. Chem. Soc., 2002, 124, 4198–4199 CrossRef CAS PubMed.
- S. J. Danishefsky and B. Simoneau, J. Am. Chem. Soc., 1989, 111, 2599–2604 CrossRef CAS.
- A. B. Jones, M. Yamaguchi, A. Patten, S. J. Danishefsky, J. A. Ragan, D. B. Smith and S. L. Schreiber, J. Org. Chem., 1989, 54, 17–20 CrossRef CAS.
- G. H. Posner, J. P. Mallamo and K. Miura, J. Am. Chem. Soc., 1981, 103, 2886–2888 CrossRef CAS.
- D. H. Hua, S. Venkataraman, R. Y. K. Chan and J. V. Paukstelis, J. Am. Chem. Soc., 1988, 110, 4741–4748 CrossRef CAS.
- G. Solladié, in Asymmetric Synthesis, ed. J. D. Morrison, Academic Press, New York, 1983 Search PubMed.
- D. H. Hua, in Advances in Carbanion Chemistry, ed. V. Snieckus, JAI Press, London, 1992 Search PubMed.
- D. H. Hua, Adv. Heterocycl. Nat. Prod. Synth., 1996, 3, 151 CAS.
- Y. Arai and T. Koizumi, Sulfur Rep., 1993, 15, 41–65 CrossRef CAS.
- P. Renaud, Chimia, 1997, 51, 236–238 CAS.
- P. Renaud and M. Gerster, Angew. Chem., Int. Ed., 1998, 37, 2562–2579 CrossRef CAS.
- J. Legros and C. Bolm, Angew. Chem., Int. Ed. Engl., 2004, 43, 4225–4228 CrossRef CAS PubMed.
- D. A. Cogan, G. Liu, K. Kim, B. J. Backes and J. A. Ellman, J. Am. Chem. Soc., 1998, 120, 8011–8019 CrossRef CAS.
- H. Egami and T. Katsuki, Synlett, 2008, 10, 1543–1546 Search PubMed.
- Z. Wang, A. Q. Peng, X. L. Sun and Y. Tang, Sci. China: Chem., 2014, 57, 1144–1149 CrossRef CAS.
- A. K. Sah and N. Baig, Catal. Lett., 2015, 145, 905–909 CrossRef CAS.
- J. Fridgen, W. A. Herrmann, G. Eickerling, A. M. Santos and F. E. Kühn, J. Organomet. Chem., 2004, 689, 2752–2761 CrossRef CAS.
- T. Fujihara, Y. Sasaki and T. Imamura, Chem. Lett., 1999, 5, 403–404 CrossRef.
- W. S. Liu, R. Zhang, J. S. Huang, C. M. Che and S. M. Peng, J. Organomet. Chem., 2001, 634, 34–38 CrossRef CAS.
- J. T. Spence and J. Tocatlian, J. Am. Chem. Soc., 1961, 83, 816–819 CrossRef CAS.
- R. Cammack, M. J. Barber and R. C. Bray, Biochem. J., 1976, 157, 469–478 CrossRef CAS PubMed.
- G. Vujevic and C. Janiak, Z. Anorg. Allg. Chem., 2003, 629, 2585–2590 CrossRef CAS.
- R. Hille, Chem. Rev., 1996, 96, 2757–2816 CrossRef CAS PubMed.
- R. H. Holm, P. Kennepohl and E. I. Solomon, Chem. Rev., 1996, 96, 2239–2314 CrossRef CAS PubMed.
- A. K. Sah, C. P. Rao, P. K. Saarenketo, E. K. Wegelius, E. Kolehmainen and K. Rissanen, Eur. J. Inorg. Chem., 2001, 2001, 2773–2781 CrossRef.
- Rigaku Corporation 2000-2010, Tokyo 196-8666, Japan.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 1997, 30, 565 CrossRef CAS.
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef CAS PubMed.
- J. Zhao, X. Zhou, A. M. Santos, E. Herdtweck, C. C. Romão and F. E. Kühn, Dalton Trans., 2003, 3736–3742 RSC.
- R. D. Chakravarthy and D. K. Chand, J. Chem. Sci., 2011, 123, 187–199 CrossRef CAS.
- K. Jeyakumar and D. K. Chand, J. Chem. Sci., 2009, 121, 111–123 CrossRef CAS.
- J. A. Schachner, P. Traar, C. Sala, M. Melcher, B. N. Harum, A. F. Sax, M. Volpe, F. Belaj and N. C. Mösch-Zanetti, Inorg. Chem., 2012, 51, 7642–7649 CrossRef CAS PubMed.
- N. Baig, G. M. Shelke, A. Kumar and A. K. Sah, Catal. Lett., 2016, 146, 333–337 CrossRef CAS.
- G. Mohammadnezhad, R. Debel and W. Plass, J. Mol. Catal. A: Chem., 2015, 410, 160–167 CrossRef CAS.
- R. L. Shriner, H. C. Struck and W. J. Jorison, J. Am. Chem. Soc., 1930, 52, 2060–2069 CrossRef CAS.
- B. Zhang, M. D. Zhou, M. Cokoja, J. Mink, S. L. Zang and F. E. Kühn, RSC. Adv., 2012, 2, 8416–8420 RSC.
- I. Sheikhshoaie, A. Rezaeifard, N. Monadi and S. Kaafi, Polyhedron, 2009, 28, 733–738 CrossRef CAS.
- R. S. Varma and K. P. Naicker, Org. Lett., 1999, 1, 189–191 CrossRef CAS.
- C. J. Carrasco, F. Montilla, E. Álvarez, C. Mealli, G. Manca and A. Galindo, Dalton Trans., 2014, 43, 13711–13730 RSC.
- K. Kamata, T. Hirano, R. Ishimoto and N. Mizuno, Dalton Trans., 2010, 39, 5509–5518 RSC.
- M. K. Panda, M. M Shaikh and P. Ghosh, Dalton Trans., 2010, 39, 2428–2440 RSC.
- R. D. Chakravarthy, K. Suresh, V. Ramkumar and D. K. Chand, Inorg. Chim. Acta, 2011, 376, 57–63 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Crystallographic data for structure S5 and SO5. CCDC 1445051 and 1445052. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra01087c |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.