
CdS-modified porous foam nickel for label-free highly efficient detection of cancer cells
Abstract
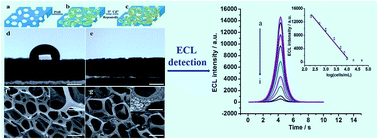
CdS-modified foam nickel (FN) was successfully constructed for the effective detection of cancer cells based on an electrochemiluminescence (ECL) technique. CdS-modified FN substrates show high ECL intensity, fast response and nice stability, which provides a new platform for the realization of an ECL sensor for the cancer cells. Two successive modification steps of 3-aminopropyltriethoxysilane (APS) and gold nanoparticles onto CdS-modified FNs not only offer substrates for conjugation of antibodies, but also effectively enhance the ECL signal, thus leading to an ECL immunosensor with high performance. Due to the large specific surface area of the FN electrodes, this biosensor, with a detection range from 200 to 10 000 cells per mL, has a detection limit of 78 cells per mL. The selectivity, stability and reproducibility of this biosensor were also demonstrated, which indicate its promising potential for clinical diagnosis.
 Please wait while we load your content...
Please wait while we load your content...

