
Merging supramolecular catalysis and aminocatalysis: amino-appended β-cyclodextrins (ACDs) as efficient and recyclable supramolecular catalysts for the synthesis of tetraketones†
Abstract
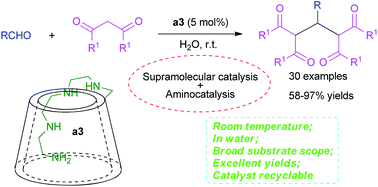
Well-designed amino-appended β-cyclodextrins (ACDs) with an amino side chain of different lengths at the primary face of β-CD were synthesized and employed in the catalytic synthesis of a series of tetraketones as supramolecular catalysts in water for the first time. Yields of 58–97% were obtained with up to 30 examples of substrate. The catalyst could be recycled easily, while a 92% yield and 84% rate of catalyst recovery could be achieved after 8 cycles of catalyst recycling. Moreover, a catalytic mechanism merging supramolecular catalysis and aminocatalysis could be proposed through detailed 1D and 2D NMR, ESI-MS and Job plot analyses. This protocol retained the promising characteristics of ambient temperature, green medium, simple operation, broad substrate scope, excellent yields, superb catalyst recycling performance and unique catalytic mechanism.
 Please wait while we load your content...
Please wait while we load your content...

