
Electrodeposition of Prussian blue films on Ni3Si2O5(OH)4 hollow nanospheres and their enhanced electrochromic properties
Abstract
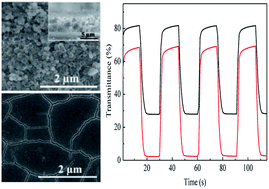
Porous Prussian blue (PB) films were successfully electrodeposited on Ni3Si2O5(OH)4 hollow nanosphere coated indium tin oxide (ITO) glass. Ni3Si2O5(OH)4 hollow nanospheres hydrothermally synthesized using SiO2 nanospheres as templates play a key role in the formation of porous PB films. The as-prepared PB films exhibit larger transmittance change, faster switching response and higher coloration efficiency than those of dense PB films directly electrodeposited on ITO glass. The enhanced electrochromic properties are attributed to the large specific surface area and porosity of the PB films which bring a high diffusion rate and short diffusion path length of ions.
 Please wait while we load your content...
Please wait while we load your content...

