
Nanoporous poly(3-hexylthiophene) thin films based on “click” prepared degradable diblock copolymers†
Abstract
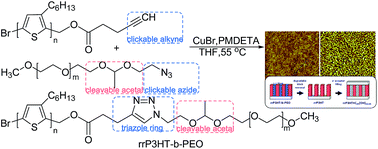
A facile approach to control the nanostructured organization of conjugated polymer platforms is proposed, based on the synthesis and characterization of copolymers containing a cleavable group inside the chain. This is illustrated by copolymerization of electroconjugated units representing regioregular poly(3-hexylthiophene) obtained by Grignard metathesis polymerization with a terminal alkynyl group and a sacrificial unit of poly(ethylene oxide) with a functionalized end azide group. The diblock copolymers were successfully synthesized via a click coupling reaction between blocks, where an acid degradable acetal group was incorporated. The strategy for formation of thin porous poly(3-hexylthiophene) films includes acid treatment of diblock copolymers, accompanied by the degradation of the acetal linker and easy removal of the poly(ethylene oxide) block. The study displays an appropriate strategy to convert nanoporous poly(3-hexylthiophene) thin films into suitable high surface area matrices, which, after successful filling with electron acceptor material, could create novel nanostructures for organic photovoltaics.
 Please wait while we load your content...
Please wait while we load your content...

