
Modulation of charge carrier mobility by side-chain engineering of bi(thienylenevinylene)thiophene containing PPE–PPVs†
Abstract
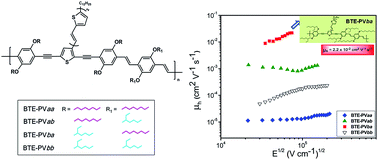
Four 2-dimensional conjugated poly(p-phenylene-ethynylene)-alt-poly(p-phenylene-vinylene) polymers containing a lateral bi(thienylenevinylene)thiophene unit (BTE-PVs) were synthesised and characterised. The investigated polymers share the same conjugated structure, but differ in the anchoring positions of solubilising linear octyloxy/branched 2-ethylhexyloxy side-chains. UV-vis spectra of the polymers in dilute chloroform solutions and as thin films were studied. X-Ray diffraction patterns as well as the bulk charge transport of polymer films cast from chlorobenzene solutions were also investigated. A dramatic effect of the solubilising side-chains on the charge carrier mobility of BTE-PV films was observed, with bulk hole mobility values ranging between 1.3 × 10−5 cm2 V−1 s−1 and 2.2 × 10−2 cm2 V−1 s−1, which is not ascribable to evident structural variations of the polymer films. It is shown that the combination of linear octyloxy and branched 2-ethylhexyloxy side-chains is favorable for the charge transport properties of BTE-PVs, compared to the incorporation of only linear or only branched side-chains.
 Please wait while we load your content...
Please wait while we load your content...

