
Deep eutectic solvent mediated synthesis of quinazolinones and dihydroquinazolinones: synthesis of natural products and drugs†
Suman Kr Ghosh and
Rajagopal Nagarajan*
School of Chemistry, University of Hyderabad, Hyderabad-500046, India. E-mail: rnsc@uohyd.ernet.in; Tel: +91 40 23134831
First published on 2nd March 2016
Abstract
A mild and greener protocol was developed to synthesize substituted quinazolinones and dihydroquinazolinones via deep eutectic solvent (DES) mediated cyclization with a series of aliphatic, aromatic, and heteroaromatic aldehydes in good to excellent yields. This greener strategy was further utilised to synthesize various quinazolinone natural products and drugs.
Introduction
Minimizing the waste and maximizing the sustainability of a reaction would be a breakthrough in the field of synthetic chemistry to favour the human race.1 Although the achievements are still limited, sustainable chemistry and its applications are at the beginning of a new era. It is very challenging for chemists to mimic nature and design an environment friendly chemical synthesis with respect to reagents, solvents and ambient conditions. Over the last decade, more efforts have been devoted towards the design of environmentally benign solvents that could be easily biodegradable or reusable.2 In recent years, green solvents like water,3 ionic liquids (ILs),4 supercritical liquids,5 and polyethylene glycol (PEG)6 have emerged to replace many organic solvents. However, the use of these solvents is restricted due to their poor solubility and the stability of organic compounds. The extensive use of ILs as solvents has been well documented for many organic reactions for over a decade but depending on their counterpart, they can be toxic and non biodegradable. Thus, nowadays the use of ILs as solvents is reduced by less than a half.7 On the other hand, deep eutectic solvents (DES) have emerged as green solvents due to their properties of polarity, low toxicity, non-volatility, biodegradability, low cost, thermal stability, and ready availability from bulk renewable resources without any further modification.8 The use of DES as a replacement for typical organic solvents in reactions like the Perkin,9 Diels–Alder,10 Heck,11 Suzuki,12 and Biginelli13 are well documented in the literature. Hence, improving on the use of DES in the synthesis of organic compounds should be the major aim.Among the nitrogen containing natural products, quinazolinone is one of the most important heterocyclic cores that accessorize many naturally occurring alkaloids as well as marketed drugs. Substituted and unsubstituted quinazolinones exhibit broad biological and pharmaceutical properties like protein tyrosine kinase inhibition, cholecystokinin inhibition, and anti-microbial, anticonvulsant, sedative, hypotensive, anti-depressant, anti-inflammatory, and anti-allergy activities.14 Certain quinazolinones which are being marketed as drugs, like methaqualone (quaalude), mebroqualone, and mecloqualone (Casfen) have sedative, hypnotic and anxiolytic properties and are used for the treatment of insomnia (Fig. 1).15 In 2011, the Wang and Che groups isolated two alkaloids, penipanoid C and 2-(4-hydroxybenzyl)quinazolin-4(3H)-one from the marine sediment-derived fungus Penicillium paneum SD-44 that exhibits cytotoxicity against the human lung carcinoma cell line A549 and BEL-7402 cell lines with IC50 values of 17.5 and 19.8 μM, respectively.16 Due to this undeniable popularity of quinazolinone alkaloids in the pharmacological industry, there is a continuous interest among researchers to develop new methods for the synthesis of the quinazolinone core. Over the years, plenty of protocols have been reported by various scientific groups using metals (Cu, Pd, Ir),17 sp3 carbon oxidation,18 multi component reactions,19 microwave conditions,20 gallium(III) triflate,21 montmorillonite K-10,22 zinc(II) perfluorooctanoate,23 KAl(SO4)2·12H2O,24 Al(H2PO4)3,25 Cu-CNTs26 and MNP-PSA (N-propylsulfamic acid supported onto magnetic Fe3O4 nanoparticles)27 as catalysts. A keen survey of the literature also reveals the use of metal free conditions like iodine,28 amberlyst-15,29 Bronsted acid,30 citric acid,31 ionic liquids,32 β-cyclodextrin,33 water–sodium dodecayl sulfate (SDS),34 cellulose-SO3H,35 CLAY,36 propane phosphonic acid anhydride (T3P)37 for the synthesis of quinazoline as well as quinazolinone. Deep eutectic solvents (DES), being more eco-friendly due to their inherent properties (vide supra), is now becoming the choice of solvent over many organic solvents. In 2012, Zhang et al. reported a three component synthesis of quinazoline in DES media.38 However, to the best of our knowledge there have been limited reports39 to date on the synthesis of quinazolinone using particularly deep eutectic solvents in spite of their aforementioned environmental impact. Furthermore, this DES mediated cyclization can be utilised for the synthesis of quinazolinone alkaloids or drugs as a key step. From an environmental perspective, the use of deep eutectic solvent mediated cyclization as a key step for the total synthesis of alkaloids would open up new directions for the scientific community. With this outlook to our ongoing research,40 herein we report a DES mediated cyclization strategy to synthesize substituted/unsubstituted quinazolinones that has been further utilised for the synthesis of various natural products and drugs.
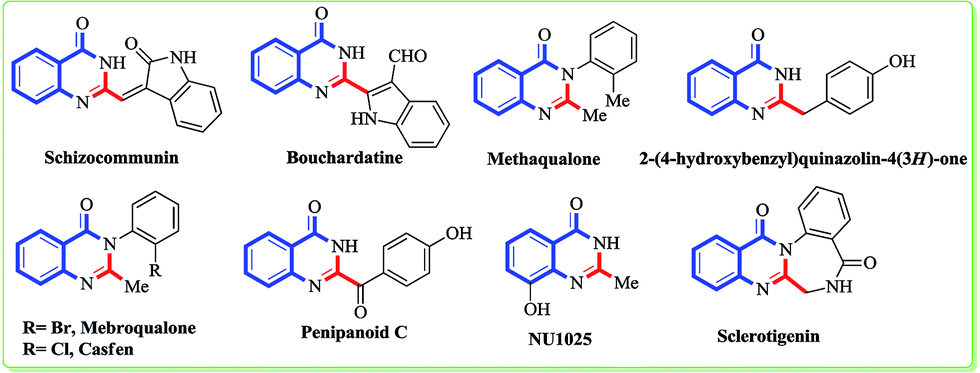 | ||
| Fig. 1 Examples of biologically and pharmaceutically important quinazolinones. | ||
Results & discussion
To obtain 2-(o-tolyl)quinazolin-4(3H)-one (3a, Scheme 1) we started our venture by optimizing a few of the deep eutectic solvent mixtures, like citric acid–N,N′-dimethylurea (DMU), D-(−)-fructose–DMU, L-(+)-tartaric acid–DMU and mannose–DMU–NH4Cl with the model substrates anthranilamide (1a) (1.0 equiv.) and o-tolualdehyde (2a) (1.2 equiv.). Among them, the L-(+)-tartaric acid–DMU (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7) mixture melt at 90 °C was found to be the most effective to give the maximum yield of compound 3a. The reaction was carried out in an open air atmosphere to aromatize the initially formed dihydroquinazoline to the quinazolinone product via aerobic oxidation. With these optimized conditions, we explored the scope and generality of the reaction using this DES. The initial formation of dihydroquinazolinone can be identified by thin layer chromatography (bright blue, long UV active) which was further aromatized to the corresponding quinazolinone via aerobic oxidation. An array of aldehydes (aromatic, aliphatic, heterocyclic) (2a–l) was exposed to these conditions with substituted/unsubstituted anthranilamides (1a–c). To our delight, all the reactions proceeded smoothly to give the corresponding dihydroquinazolinone/quinazolinone depending on the time of the reaction. The electron donating aromatic/aliphatic aldehydes and anthranilamides were converted smoothly to the cyclized quinazolinone product within 6–15 h in good yields (3a, 3b, 3d), whereas, in case of benzaldehyde we obtained both the quinazolinone (3c) and dihydroquinazolinone (3c′) product even after 24 h. The electron withdrawing aromatic aldehydes were not converted to the corresponding quinazolinone after 24 h, although we had obtained the corresponding dihydro derivatives (3e′ and 3h′) within 2 h of the reaction.
:7) mixture melt at 90 °C was found to be the most effective to give the maximum yield of compound 3a. The reaction was carried out in an open air atmosphere to aromatize the initially formed dihydroquinazoline to the quinazolinone product via aerobic oxidation. With these optimized conditions, we explored the scope and generality of the reaction using this DES. The initial formation of dihydroquinazolinone can be identified by thin layer chromatography (bright blue, long UV active) which was further aromatized to the corresponding quinazolinone via aerobic oxidation. An array of aldehydes (aromatic, aliphatic, heterocyclic) (2a–l) was exposed to these conditions with substituted/unsubstituted anthranilamides (1a–c). To our delight, all the reactions proceeded smoothly to give the corresponding dihydroquinazolinone/quinazolinone depending on the time of the reaction. The electron donating aromatic/aliphatic aldehydes and anthranilamides were converted smoothly to the cyclized quinazolinone product within 6–15 h in good yields (3a, 3b, 3d), whereas, in case of benzaldehyde we obtained both the quinazolinone (3c) and dihydroquinazolinone (3c′) product even after 24 h. The electron withdrawing aromatic aldehydes were not converted to the corresponding quinazolinone after 24 h, although we had obtained the corresponding dihydro derivatives (3e′ and 3h′) within 2 h of the reaction.
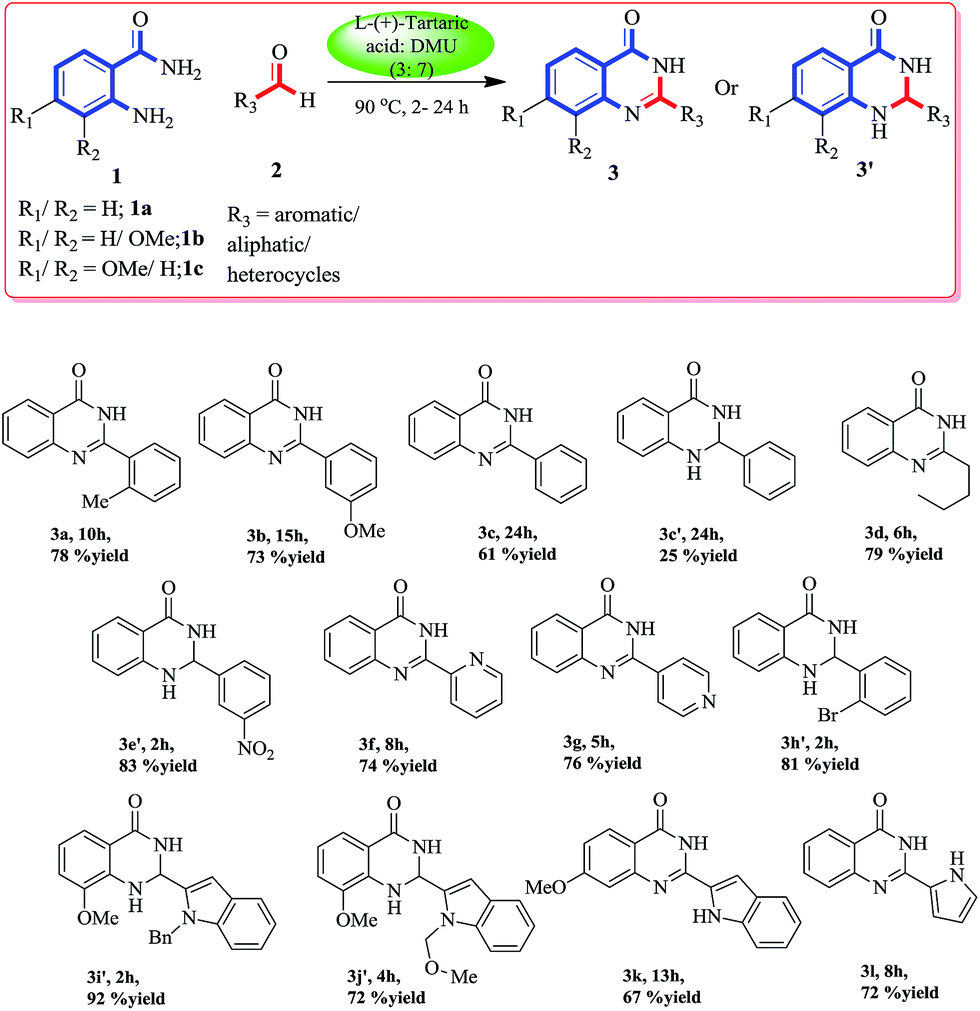 | ||
| Scheme 1 Synthesis of 2-substituted quinazolinonea. aReaction was performed with 1 (1.0 equiv.) and 2 (1.2 equiv.), in L-(+)-tartaric acid:DMU (3:7) mixture at 90 °C in an open mouth round bottomed flask. | ||
The same outcome was obtained when we used substituted anthranilamides (1b and 1c) with heterocyclic (pyridine, indole, and pyrrole) aldehydes. This observation showed that with the variation of time, both dihydroquinazolinone products as well as quinazolinone products can be obtained. All the compounds were extracted with ethyl acetate and purified using column chromatography. Once we gained an insight into this DES mediated cyclization, to synthesize various substituted dihydroquinazolinones and quinazolinones, we shifted our focus to apply this protocol to the formal synthesis of some biologically important quinazolinone natural products and drugs.
Bouchardatine41 (Fig. 1), is a very well known β-indoloquinazoline alkaloid isolated from B. neurococca (Rutaecae), showing wide biological properties like anti-cancer, anti-inflammatory and anti-tuberculosis activity. Recently it has also been documented as a novel inhibitor of adipogenesis/lipogenesis in 3T3-L1 adipocytes. Due to these vital properties, it immediately drew our attention and we were eager to apply our DES mediated protocol to synthesizing it. Indole-2-carboxaldehyde (4) was obtained in two steps from indole-2-carboxylic acid which on treatment with anthranilamide (1a) in L-(+)-tartaric acid–DMU (3:7) melt at 90 °C for 8 h gave 2-(1H-indol-2-yl)quinazolin-4(3H)-one (5) in 81% yield. Formylation of compound 5 with DMF/POCl3 will introduce the aldehyde group at indole C3 to give bouchardatine, which has already been reported by our group (Scheme 2, part A).42
 | ||
| Scheme 2 Formal synthesis of 2-substituted quinazolinone alkaloids. | ||
In part B, a formal synthesis of schizocommunin was achieved. Schizocommunin was first reported in 1999 from the liquid culture medium of Schizophyllum commune by Hosoe et al. It shows strong cytotoxic activity against murine lymphoma cells. Further biological studies on schizocommunin were prevented because of its scarcity from natural sources, and there had been no reports of the total synthesis of schizocommunin until Nishida et al. in 2013 reported the revised structure.43 Their synthesis of schizocommunin was demonstrated from 2-methyl-4(3H)-quinazolinone on refluxing in acetic acid media with isatin. Hence, the scarcity of schizocommunin in nature, and the high cost of the commercially available 2-methyl-4(3H)-quinazolinone attracted us. 2-Methyl-4(3H)-quinazolinone could be easily prepared via our protocol from the cheaper starting material anthranilamide (1a). Hence, anthranilamide (1.0 equiv.) was treated with acetaldehyde (3.0 equiv.) in the green melt at 90 °C for 2 h to obtain the corresponding dihydroquinazolinone (7′) which was further treated with 2,3-dichloro-5,6-dicyano-p-benzoquinone (DDQ) in dry DCM at rt to acquire 2-methyl-4(3H)-quinazolinone (7) in 85% yield (Scheme 2, part B). Schizocommunin can be synthesized in one step from compound 7 following Nishida’s procedure.
Though 2,2′-disubstituted quinazolinones are not found often in naturally occurring alkaloids, their synthetic versions show a wide spectrum of pharmacological activities. The incorporation of a spirocyclohexane, aliphatic or hetrocyclic moiety at C2 of the quinazolinone heterocycle gives anti-inflammatory, analgesic, and antileishmanial agents. Some spiroquinazolinones exhibiting anti-amoebic activity in vitro were also tested as central nervous system depressants.44 Thus we were excited to test our protocol for the synthesis of different 2,2′-disubstituted quinazolinones. A variety of ketones reacted smoothly over the melt at 90 °C with anthranilamide (1a) to furnish the corresponding substituted quinazolinones. A wide range of compounds (aliphatic–aliphatic, aliphatic–aromatic, aromatic–aromatic, cyclic ketones and isatin) was well tolerated under these reaction conditions to give excellent yields of the products (Scheme 3).
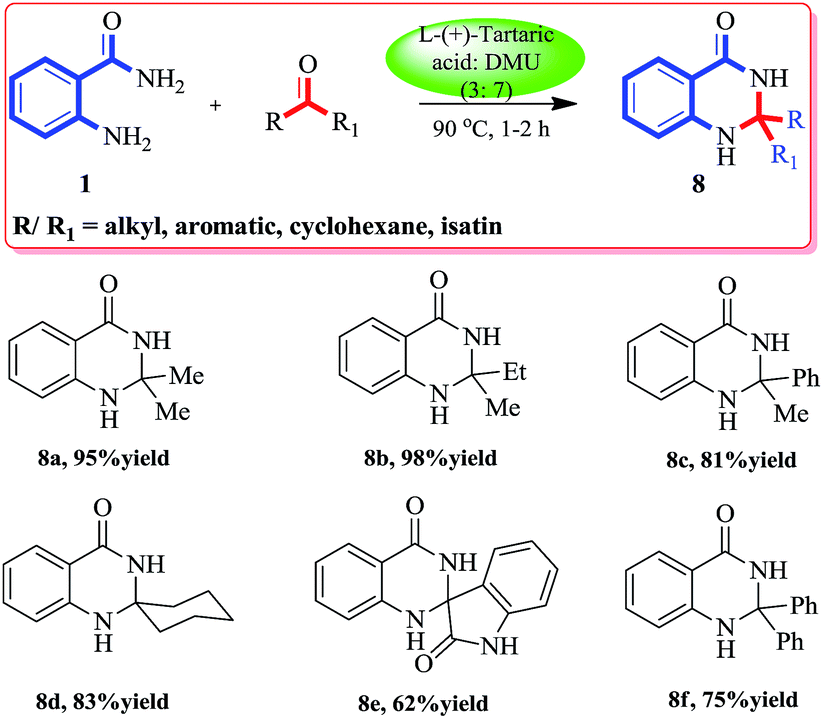 | ||
| Scheme 3 Synthesis of 2,2′-disubstituted quinazolinonesb. bReaction was performed with 1a (1.0 equiv.) and the ketone (1.0 equiv.), in a L-(+)-tartaric acid:DMU (3:7) mixture at 90 °C in an open mouth round bottomed flask. | ||
After the successful manifestation of our green protocol for 2-substituted/2,2-disubstituted quinazolinones, we were eager to explore the scope of the reaction on the synthesis of 2,3-disubstituted quinazolinones. The significance of this quinazolinone core is more prominent, particularly in respect to its pharmacological properties (vide supra, Fig. 1).
Hence, we selected some of the drugs (methaqualone, mecloqualone, and mebroqualone) and alkaloids as target molecules to validate our protocol.
To synthesize the exact target molecules, we prepared the corresponding starting material 2-amino-N-(substituted) benzamide from 2-nitrobenzoic acid in two steps. In the first step, 2-nitrobenzoic acid was stirred with oxalyl chloride in dry DCM for 3 h at rt, followed by the removal of excess solvent and oxalyl chloride under pressure, producing the corresponding acid chloride. Next, this acid chloride was added to the mixture of substituted benzamides and triethylamine, and stirred for another 4–6 h at rt to furnish the respective 2-nitro-N-(substituted)benzamides (9a–e).45 Nitro group reduction of compounds 9a–e in the presence of Zn/NH4Cl gave the 2-amino-N-(substituted)benzamides (10a–e) in good yields. Pleasingly, all of the 2-amino-N-(substituted)benzamides reacted smoothly with the corresponding aldehydes in the green melt at 90 °C to give the corresponding dihydroquinazolinones (11a–f) in good to excellent yield.
Meanwhile in the NMR spectrum, we observed the firm existence of two isomers for compounds 11a, 11b, 11c and 11e (see ESI†) with different ratios. We presumed that the presence of the R1 group in compound 11 restricts the C–C bond rotation and generates conformational isomerism. To prove the existence of rotamers, we carried out a temperature variant NMR experiment where spectral changes were measured in DMSO-d6 solvent over a temperature range of RT-80 °C. As Fig. 2 for compound 11b shows, with the increase in temperature the signal spread (Δδ) between the signals of peaks a (3H, s) and b (1H, q) decreases. At 50 °C the signals of a and b start to coalesce on the NMR time scale. The measurement at 80 °C shows that the peaks of both a and b merged as both the conformers lost their identity and exhibited a rapidly equilibrating species.
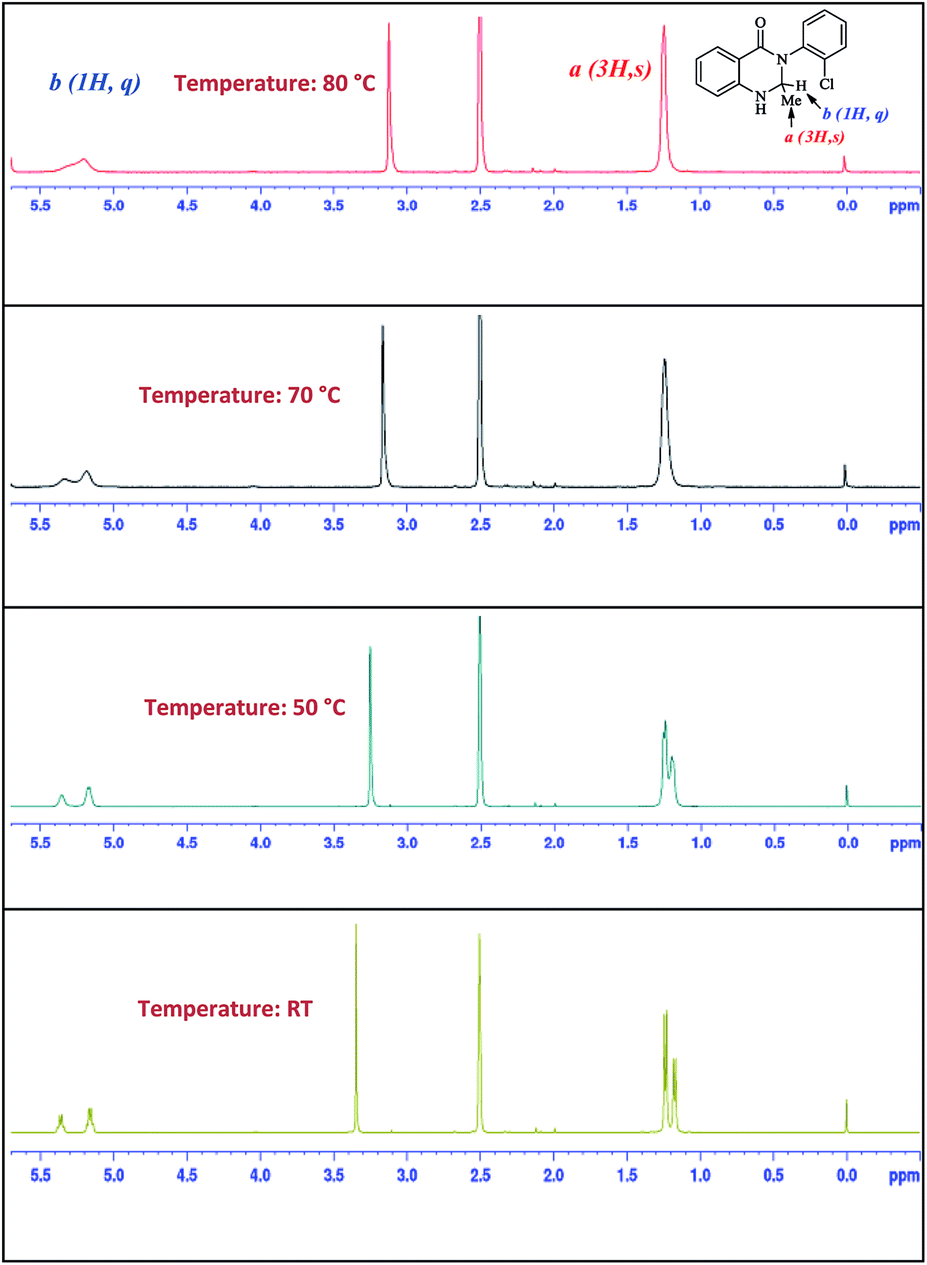 | ||
| Fig. 2 Temperature effect on the conformational isomerism of 11b. | ||
Next, the dihydroquinazolinones were aromatized with DDQ to furnish the respective quinazolinones in good yields (12a–f). We have tested a variety of substitutes on aniline, particularly halo, ester, and alkyl, to synthesis the exact drugs and alkaloids. To our delight, all the reactions proceeded with ease to give excellent yields of the compounds including 2-(4-methoxyphenyl)-3-phenylquinazolin-4(3H)-one (12f). Drugs like methaqualone (12a), mecloqualone (12b), mebroqualone (12c) and the alkaloid methyl 2-(2-methyl-4-oxoquinazolin-3(4H)-yl)benzoate (12e) were synthesized with good overall yields (Scheme 4). Compounds 12a and 12e can be further converted to two more natural products, piriqualone and sclerotigenin, in one and two steps via Welch46 and Zhou’s47 method (Scheme 5).
 | ||
| Scheme 4 Synthesis of 2,3 substituted quinazolinones (drugs and natural products). | ||
 | ||
| Scheme 5 Formal syntheses of sclerotigenin and piriqualone. | ||
Conclusions
In conclusion, we have developed a greener and cheaper strategy to synthesise substituted and unsubstituted dihydroquinazolinones as well as quinazolinones in good to excellent yields. Numerous aldehydes/ketones and substituted anthranilamides were well tolerated under these optimized conditions. This protocol was further utilized for the formal synthesis of many naturally occurring alkaloids and drugs.Experimental section
General information
1H and 13C-NMR spectra were recorded at 400 and 100 MHz, respectively, or at 500 and 125 MHz, respectively. Chemical shifts were calculated in ppm downfield from TMS (δ = 0) for 1H NMR, and relative to the central CDCl3 resonance (δ = 77.0) and DMSO-d6 (δ = 39.51) for 13C NMR. Data presented in the experimental section are as follows: chemical shift, integration, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, dd = doublet doublet), coupling constant in hertz (Hz). TOF and quadrupole mass analyzer types were used for the HRMS measurements. Mass spectral data was obtained from HRMS (ESI). IR spectra were recorded using a FT-IR spectrometer using KBr pellets or neat. Melting points were measured in open capillary tubes and are uncorrected. All the obtained products were purified by column chromatography using silica gel (100–200 mesh). All reaction solvents used were dried from GR grade solvents. All other commercial reagents were used as received.General experimental procedure to synthesize dihydroquinazolinone/quinazolinones
For 0.100 g of starting amide, a total 2 g mixture of L-(+)-tartaric acid and N,N′-dimethylurea (DMU) in a ratio of 3:7 was taken in an oven dried open mouth round bottomed flask and heated to its eutectic point of 70 °C, where the mixture melted to give a clear solution. Next, the aldehyde (1.2/1.5 equiv.) and substituted anthranilamide (1.0 equiv.) were added to this melt and heated at 90 °C for 2–24 h, depending on the desired product. On completion of the reaction (checked by thin layer chromatography), water was added. The mixture was extracted with EtOAc, dried over Na2SO4 and evaporated under vacuum. The residue was purified by column chromatography on silica gel using an ethyl acetate/hexane mixture to afford the desired dihydroquinazolinone/quinazolinone.
:hexanes = 3:7); IR (KBr) 3057, 1670, 1601, 1469, 1286, 1264, 765 cm−1; 1H NMR (400 MHz, CDCl3) δ 11.49 (1H, s, br), 8.34 (1H, d, J = 7.6 Hz), 8.15 (2H, d, J = 8.0 Hz), 7.82 (2H, t, J = 8.0 Hz), 7.50 (1H, t, J = 8.0 Hz), 7.38 (2H, d, J = 2.0 Hz), 2.47 (3H, s); 13C NMR (100 MHz, CDCl3) δ 164.1, 151.9, 149.6, 142.1, 134.8, 130.0, 129.7, 127.9, 127.4, 127.3, 126.5, 126.3, 120.8, 21.5; m/z = 237 (M + H), positive mode; anal. calcd for C15H12N2O: C, 76.25; H, 5.12; N, 11.86%. Found: C, 76.12; H, 5.18; N, 11.96%.:hexanes = 3:7); IR (KBr) 1678, 1602, 1484, 1264, 834 cm−1; 1H NMR (500 MHz, DMSO-d6) δ 12.39 (1H, s, br), 8.19 (2H, d, J = 8.5 Hz), 8.13 (1H, d, J = 7.0 Hz), 7.81 (1H, t, J = 6.5 Hz), 7.70 (1H, d, J = 8.0 Hz), 7.48 (1H, t, J = 7.0 Hz), 7.09 (2H, d, J = 9.0 Hz), 3.85 (3H, s); 13C NMR (125 MHz, DMSO-d6) δ 162.7, 162.4, 152.3, 149.4, 134.9, 129.9, 127.7, 126.5, 126.3, 125.3, 121.2, 114.5, 55.9; m/z = 253 (M + H), positive mode; anal. calcd for C15H12N2O2: C, 71.42; H, 4.79; N, 11.10%. Found: C, 71.57; H, 4.71; N, 11.26%.:hexanes = 3:7); IR (KBr) 2918, 1666, 1601, 1291, 1265, 768 cm−1; 1H NMR (400 MHz, CDCl3 + DMSO-d6) δ 11.94 (1H, s), 8.05 (1H, d, J = 7.2 Hz), 7.99–7.98 (2H, m), 7.57–7.55 (2H, m), 7.32–7.29 (3H, m), 7.27–7.21 (1H, m); 13C NMR (100 MHz, CDCl3 + DMSO-d6) δ 163.2, 152.3, 149.2, 134.3, 132.9, 131.2, 129.5, 128.6, 128.1, 127.6, 126.3, 126.0, 121.1; m/z = 223 (M + H), positive mode; anal. calcd for C14H10N2O: C, 75.66; H, 4.54; N, 12.60%. Found: C, 75.81; H, 4.51; N, 12.53%.:hexanes = 3:7); IR (KBr) 3299, 2925, 1651, 1608, 1264 cm−1; 1H NMR (400 MHz, DMSO-d6) δ 8.29 (1H, s), 7.63 (1H, d, J = 7.6 Hz), 7.51 (2H, d, J = 7.6 Hz), 7.42–7.34 (3H, m), 7.26 (1H, t, J = 7.2 Hz), 7.12 (1H, s), 6.77 (1H, d, J = 8.4 Hz), 6.69 (1H, t, J = 7.2 Hz), 5.77 (1H, s); 13C NMR (100 MHz, DMSO-d6) δ 164.1, 148.3, 142.1, 133.8, 128.9, 128.8, 127.8, 127.3, 117.6, 115.4, 114.9, 67.0; m/z = 225 (M + H), positive mode; anal. calcd for C14H12N2O: C, 74.98; H, 5.39; N, 12.49%. Found: C, 74.82; H, 5.45; N, 12.56%.:hexanes = 3:7); IR (KBr) 2919, 1682, 1616, 1467, 1261, 1130 cm−1; 1H NMR (400 MHz, DMSO-d6) δ 12.02 (1H, s), 8.29 (1H, d, J = 8.0 Hz), 7.77 (1H, t, J = 8.0 Hz), 7.71 (1H, d, J = 8.0 Hz), 7.47 (1H, t, J = 8.0 Hz), 2.81 (2H, t, J = 8.0 Hz), 1.92–1.82 (2H, m), 1.51 (2H, q, J = 7.4 Hz), 1.00 (3H, t, J = 8.0 Hz); 13C NMR (100 MHz, DMSO-d6) δ 164.4, 157.0, 149.5, 134.8, 127.2, 126.3, 126.2, 120.5, 35.7, 29.7, 22.4, 13.8; HRMS (ESI-MS) calcd for C12H14N2O (M + H) 203.1184, found 203.1187.:hexanes = 3:7); IR (KBr) 2920, 1731, 1682, 1616, 1468, 1200, 1145 cm−1; 1H NMR (400 MHz, DMSO-d6) δ 8.53 (1H, s), 8.35 (1H, s), 8.19 (1H, d, J = 8.0 Hz), 7.93 (1H, d, J = 7.2 Hz), 7.68 (1H, t, J = 8.0 Hz), 7.61 (1H, d, J = 7.2 Hz), 7.34 (1H, s), 7.26 (1H, t, J = 7.2 Hz), 6.78 (1H, d, J = 8.0 Hz), 6.68 (1H, t, J = 7.2 Hz), 5.94 (1H, s); 13C NMR (100 MHz, DMSO-d6) δ 163.8, 148.2, 147.8, 144.7, 134.1, 133.8, 130.5, 127.9, 123.7, 122.0, 118.0, 115.4, 115.1, 65.6; HRMS (ESI-MS) cald for C14H11N3O3 (M + H) 270.0878, found 270.0874.:hexanes = 3:7); IR (KBr) 1684, 1607, 1472, 1331, 769 cm−1; 1H NMR (400 MHz, DMSO-d6) δ 11.82 (1H, s, br), 8.74 (1H, d, J = 4.4 Hz), 8.43 (1H, d, J = 8.0 Hz), 8.17 (1H, d, J = 8.0 Hz), 8.06 (1H, t, J = 8.0 Hz), 7.86 (1H, t, J = 8.0 Hz), 7.79 (1H, d, J = 8.0 Hz), 7.65–7.62 (1H, m), 7.56 (1H, t, J = 7.6 Hz); 13C NMR (100 MHz, DMSO-d6) δ 161.3, 150.3, 149.5, 149.0, 148.8, 138.5, 135.2, 128.1, 127.8, 127.0, 126.5, 122.6, 122.4; m/z = 224 (M + H), positive mode; anal. calcd for C13H9N3O: C, 69.95; H, 4.06; N, 18.82%. Found: C, 69.86; H, 4.15; N, 18.75%.:hexanes = 1:1); IR (KBr) 3358, 2964, 1682, 1561, 1468, 1260, 1030 cm−1; 1H NMR (500 MHz, DMSO-d6) δ 12.77 (1H, s), 8.79 (2H, d, J = 4.0 Hz), 8.18 (1H, d, J = 7.6 Hz), 8.12 (2H, d, J = 4.5 Hz), 7.88 (1H, t, J = 7.5 Hz), 7.79 (1H, d, J = 7.9 Hz), 7.58 (1H, t, J = 7.2 Hz); 13C NMR (125 MHz, DMSO-d6) δ 162.5, 151.0, 150.7, 140.4, 135.3, 128.2, 127.9, 126.4, 122.0, 121.9; HRMS (ESI-MS) calcd for C13H9N3O (M + H) 224.0824, found 224.0820.:hexanes = 3:7); IR (KBr) 3220, 1656, 1612, 1486, 1249 cm−1; 1H NMR (400 MHz, DMSO-d6) δ 8.21 (1H, s), 7.69–7.65 (3H, m), 7.44 (1H, t, J = 7.2 Hz), 7.32 (1H, t, J = 7.6 Hz), 7.27 (1H, t, J = 7.6 Hz), 7.00 (1H, s), 6.78 (1H, d, J = 8.4 Hz), 6.74 (1H, t, J = 7.6 Hz), 6.11 (1H, s); 13C NMR (100 MHz, DMSO-d6) δ 164.2, 148.1, 139.6, 133.9, 133.3, 131.2, 129.5, 128.5, 127.9, 122.6, 118.0, 115.1, 115.0, 66.8; HRMS (ESI-MS) calcd for C14H11BrN2O (M + Na) 324.9952, found 324.9959.:hexanes = 3:7); IR (KBr) 2920, 1671, 1610, 1457, 1249 cm−1; 1H NMR (400 MHz, DMSO-d6) δ 8.48 (1H, s), 7.52 (1H, d, J = 7.6 Hz), 7.33–7.28 (5H, m), 7.09–6.94 (5H, m), 6.69 (1H, t, J = 7.6 Hz), 6.43 (1H, s, br), 6.39 (1H, s), 6.05 (1H, s), 5.65 (2H, s), 3.72 (3H, s); 13C NMR (100 MHz, DMSO-d6) δ 164.0, 146.9, 140.5, 138.6, 137.8, 136.5, 129.0, 127.6, 127.0, 126.8, 122.2, 120.9, 120.0, 119.3, 117.4, 116.0, 114.0, 110.8, 101.9, 60.1, 56.0, 46.7; HRMS (ESI-MS) calcd for C24H21N3O2 (M + H) 384.1712, found 384.1706.:hexanes = 3:7); IR (KBr) 3419, 1687, 1605, 1468, 1276, 761 cm−1; 1H NMR (400 MHz, CDCl3 + DMSO-d6) δ 7.57 (1H, s, br), 7.52–7.43 (3H, m), 7.22 (1H, t, J = 8.0 Hz), 7.09 (1H, t, J = 8.0 Hz), 6.85 (1H, dd, J = 1.2 Hz, J = 8.0 Hz), 6.74 (1H, t, J = 8.0 Hz), 6.63 (1H, s), 6.16 (1H, d, J = 2.0 Hz), 5.70 (1H, s, br), 5.66 (1H, d, J = 11.2 Hz), 5.55 (1H, d, J = 11.6 Hz), 3.79 (3H, s), 3.31 (3H, s); 13C NMR (100 MHz, CDCl3 + DMSO-d6) δ 169.5, 151.2, 143.3, 142.7, 141.8, 131.7, 127.8, 125.9, 125.4, 124.2, 122.8, 120.2, 118.3, 114.2, 108.9, 78.9, 66.3, 60.8, 60.4; m/z = 338 (M + H), positive mode; anal. calcd for C19H19N3O3: C, 67.64; H, 5.68; N, 12.46%. Found: C, 67.49; H, 5.73; N, 12.38%.:hexanes = 3:7); IR (KBr) 3484, 2898, 1654, 1600, 1315, 1145 cm−1; 1H NMR (400 MHz, DMSO-d6) δ 12.48 (1H, s), 11.77 (1H, s), 8.06 (1H, t, J = 9.2 Hz), 7.63 (2H, d, J = 11.2 Hz), 7.51 (1H, d, J = 8.4 Hz), 7.21 (1H, t, J = 8.4 Hz), 7.12 (1H, s), 7.09–7.03 (2H, m), 3.91 (3H, s); 13C NMR (100 MHz, DMSO-d6) δ 164.6, 161.8, 151.4, 147.6, 138.1, 130.5, 128.2, 127.9, 124.5, 122.0, 120.4, 116.1, 115.0, 112.9, 108.6, 105.4, 56.1; HRMS (ESI-MS) calcd for C17H13N3O2 (M + H) 292.1086, found 292.1086.:hexanes = 3:7); IR (KBr) 2919, 1672, 1596, 1496, 1261, 765 cm−1; 1H NMR (400 MHz, DMSO-d6) δ 12.18 (1H, s), 11.70 (1H, s), 8.08 (1H, d, J = 7.6 Hz), 7.77 (1H, t, J = 7.6 Hz), 7.62 (1H, d, J = 8.0 Hz), 7.41 (1H, t, J = 7.2 Hz), 7.29 (1H, s), 7.05 (1H, s), 6.21 (1H, s); 13C NMR (100 MHz, DMSO-d6) δ 162.4, 149.7, 146.8, 135.0, 126.9, 126.4, 125.7, 124.7, 124.3, 120.9, 112.9, 110.2; m/z = 212 (M + H), positive mode; anal. calcd for C12H9N3O: C, 68.24; H, 4.29; N, 19.89%. Found: C, 68.15; H, 4.36; N, 19.78%.:hexanes = 1:1); IR (KBr) 3413, 1665, 1589, 1468, 1260, 772 cm−1; 1H NMR (400 MHz, DMSO-d6) δ 12.62 (1H, s), 11.81 (1H, s), 8.17 (1H, d, J = 8.0 Hz); 7.88–7.85 (1H, m), 7.75 (1H, d, J = 8.0 Hz), 7.68 (1H, s), 7.65 (1H, d, J = 8.0 Hz), 7.54 (2H, t, J = 7.2 Hz), 7.24 (1H, t, J = 8.4 Hz), 7.07 (1H, t, J = 7.2 Hz); 13C NMR (125 MHz, DMSO-d6) δ 162.3, 149.2, 147.0, 138.1, 135.2, 130.5, 127.9, 127.4, 126.7, 126.5, 124.5, 122.0, 121.6, 120.4, 112.9, 105.5; HRMS (ESI-MS) calcd for C16H11N3O (M + H) 262.0980, found 262.0980.:hexanes = 1:1); IR (KBr) 3266, 1668, 1615, 1257, 753 cm−1; 1H NMR (400 MHz, DMSO-d6) δ 7.90 (1H, s), 7.59 (1H, d, J = 8.0 Hz), 7.23 (1H, t, J = 8.0 Hz), 6.68 (2H, d, J = 8.0 Hz), 6.60 (1H, s), 4.82 (1H, q, J = 5.6 Hz), 1.31 (3H, d, J = 5.6 Hz); 13C NMR (100 MHz, DMSO-d6) δ 164.6, 149.2, 133.6, 127.9, 117.6, 115.6, 114.8, 61.3, 21.7; HRMS (ESI-MS) calcd for C9H10N2O (M + H) 163.0871, found 163.0870.:hexane) to afford the desired product (7). This compound was obtained as a yellowish brown crystalline solid; yield = 0.042 g/0.050 g of 7′, 85%; mp = 228 °C; Rf = 0.21 (EtOAc:hexanes = 1:1); IR (KBr) 2915, 1693, 1665, 1610, 1468, 1254 cm−1; 1H NMR (400 MHz, CDCl3) δ 11.81 (1H, s, br), 8.29 (1H, d, J = 8.0 Hz), 7.78 (1H, t, J = 8.0 Hz), 7.69 (1H, d, J = 8.0 Hz), 7.48 (1H, t, J = 8.0 Hz), 2.60 (3H, s); 13C NMR (100 MHz, CDCl3) δ 164.2, 153.2, 149.4, 134.9, 127.0, 126.4, 126.2, 120.3, 22.1; HRMS (ESI-MS) calcd for C9H8N2O (M + H) 161.0715, found 161.0710; we followed the same procedure for compounds 12a–f.:hexanes = 1:1); IR (KBr) 3255, 1632, 1485, 1270, 1175, 750 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.89 (1H, d, J = 7.2 Hz), 7.31 (1H, t, J = 8.0 Hz), 7.05 (1H, s, br), 6.83 (1H, t, J = 7.2 Hz), 6.64 (1H, d, J = 7.6 Hz), 4.25 (1H, s, br), 1.58 (6H, s); 13C NMR (100 MHz, CDCl3) δ 164.7, 146.0, 133.9, 128.3, 118.7, 114.7, 114.6, 67.6, 29.6; m/z = 177 (M + H), positive mode. Anal. calcd for C10H12N2O: C, 68.16; H, 6.86; N, 15.90%. Found: C, 68.26; H, 6.81; N, 15.82%.:hexanes = 1:1); IR (KBr) 3270, 1638, 1612, 1266, 754 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.88 (1H, d, J = 7.5 Hz), 7.31 (1H, d, J = 8.5 Hz), 6.82 (1H, t, J = 7.0 Hz), 6.62 (1H, d, J = 8.0 Hz), 6.17 (1H, s, br), 4.13 (1H, s, br), 1.82 (2H, q, J = 7.4 Hz), 1.51 (3H, s), 1.01 (3H, t, J = 7.4 Hz); 13C NMR (100 MHz, CDCl3) δ 165.1, 146.1, 134.1, 128.3, 118.4, 114.5, 114.2, 70.1, 34.8, 27.5, 8.2; m/z = 191 (M + H), positive mode. Anal. calcd for C11H14N2O: C, 69.45; H, 7.42; N, 14.73%. Found: C, 69.56; H, 7.35; N, 14.62%.:hexanes = 3:7); IR (KBr) 3402, 1665, 1610, 1501, 1380, 1265 cm−1; 1H NMR (500 MHz, CDCl3 + DMSO-d6) δ 7.64 (1H, d, J = 8.0 Hz), 7.60 (1H, s, br), 7.43 (2H, d, J = 8.0 Hz), 7.17 (2H, t, J = 8.0 Hz), 7.14–7.08 (2H, m), 6.64 (1H, d, J = 8.0 Hz), 6.59 (1H, t, J = 8.0 Hz), 5.98 (1H, s, br), 1.72 (3H, s); 13C NMR (100 MHz, CDCl3 + DMSO-d6) δ 164.8, 146.4, 146.0, 133.7, 128.2, 128.0, 127.6, 125.2, 118.1, 115.2, 114.7, 70.7, 30.3; HRMS (ESI-MS) calcd for C15H14N2O (M + Na) 261.1004, found 261.1003.:hexanes = 3:7); IR (KBr) 3169, 2922, 2852, 1642, 1606, 1479, 1267 cm−1; 1H NMR (400 MHz, CDCl3 + DMSO-d6) δ 7.62–7.58 (1H, m), 7.06 (1H, d, J = 6.8 Hz), 6.89 (1H, s), 6.55–6.52 (2H, m), 5.21 (1H, s, br), 1.63 (4H, s), 1.41 (4H, s), 1.26 (2H, s); 13C NMR (100 MHz, CDCl3 + DMSO-d6) δ 164.3, 146.2, 133.5, 127.8, 117.7, 114.7, 68.2, 37.4, 24.6, 21.6; m/z = 217 (M + H), positive mode. Anal. calcd for C13H16N2O: C, 72.19; H, 7.46; N, 12.95%. Found: C, 72.25; H, 7.41; N, 13.07%.:hexanes = 1:1); IR (KBr) 1726, 1659, 1609, 1585, 1264 cm−1; 1H NMR (400 MHz, DMSO-d6) δ 10.31 (1H, s), 8.36 (1H, s), 7.61 (1H, d, J = 7.2 Hz), 7.48 (1H, d, J = 7.2 Hz), 7.34 (1H, t, J = 7.6 Hz), 7.28 (1H, s), 7.24 (1H, t, J = 8.0 Hz), 7.07 (1H, t, J = 7.6 Hz), 6.87 (1H, d, J = 7.6 Hz), 6.69 (1H, t, J = 7.6 Hz), 6.62 (1H, d, J = 7.6 Hz); 13C NMR (100 MHz, DMSO-d6) δ 176.5, 164.5, 147.3, 142.5, 133.8, 131.3, 129.8, 127.3, 125.8, 122.8, 117.7, 114.7, 114.3, 110.6, 71.4; m/z = 266 (M + H), positive mode; anal. calcd for C15H11N3O2: C, 67.92; H, 4.18; N, 15.84%. Found: C, 67.85; H, 4.12; N, 15.76%.:hexanes = 3:7); IR (KBr) 3375, 3243, 1649, 1610, 1484, 1375, 1210, 750 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.81 (1H, d, J = 7.6 Hz), 7.43–7.40 (4H, m), 7.34–7.27 (7H, m), 6.78 (1H, t, J = 7.6 Hz), 6.73 (2H, d, J = 8.0 Hz), 5.36 (1H, s, br); 13C NMR (125 MHz, CDCl3) δ 164.3, 145.6, 143.7, 134.2, 128.6, 128.5, 127.3, 119.2, 115.4, 114.9, 76.0; HRMS (ESI-MS) calcd for C20H16N2O (M + Na) 323.1160, found 323.1163.:1, 20 mL). To this solution, NH4Cl (0.353 g, 6.6 mmol) and Zn dust (1.15 g, 17.6 mmol) were added. The reaction was stirred at rt for 5–6 h. After completion of the reaction, the reaction mixture was filtered and extracted with ethyl acetate and evaporated to dryness. The residue was purified by column chromatography on silica gel to give compound 10a. We followed the same procedure for the preparation of compounds 10b–e. This compound was obtained as a yellowish white solid; yield = 0.380 g/0.500 g of 10a, 86%; mp = 104 °C; Rf = 0.55 (EtOAc:hexanes = 3:7); 1H NMR (400 MHz, CDCl3) δ 7.84 (1H, d, J = 8.0 Hz), 7.66 (1H, s, br), 7.53 (1H, d, J = 8.0 Hz), 7.31–7.26 (3H, m), 7.15 (1H, t, J = 7.2 Hz), 6.76 (2H, d, J = 8.0 Hz), 5.56 (2H, s, br), 2.35 (3H, s); 13C NMR (100 MHz, CDCl3) δ 167.6, 149.2, 135.7, 132.8, 130.6, 130.0, 127.2, 126.8, 125.5, 123.7, 117.6, 116.8, 116.1, 17.9; HRMS (ESI-MS) calcd for C14H14N2O (M + Na) 249.1004, found 249.1008.:hexanes = 3:7); 1H NMR (400 MHz, CDCl3) δ 8.48 (1H, d, J = 8.4 Hz), 8.37 (1H, s, br), 7.57 (1H, d, J = 7.6 Hz), 7.44 (1H, d, J = 8.0 Hz), 7.37–7.29 (2H, m), 7.10 (1H, t, J = 8.0 Hz), 6.77 (2H, t, J = 7.6 Hz), 5.63 (2H, s, br); 13C NMR (100 MHz, CDCl3) δ 167.3, 149.4, 134.8, 133.1, 129.1, 127.7, 127.2, 124.6, 123.4, 121.7, 117.7, 116.9, 115.6; HRMS (ESI-MS) calcd for C13H11ClN2O (M + H) 247.0638, found 247.0636.:hexanes = 3:7); 1H NMR (400 MHz, CDCl3) δ 8.47 (1H, dd, J = 1.6 Hz, J = 8.4 Hz), 8.37 (1H, s, br), 7.60 (2H, t, J = 7.6 Hz), 7.39 (1H, t, J = 7.6 Hz), 7.33–7.29 (1H, m), 7.03 (1H, dt, J = 1.2 Hz, J = 7.6 Hz), 6.77 (2H, t, J = 8.0 Hz), 5.65 (2H, s, br); 13C NMR (100 MHz, CDCl3) δ 169.3, 149.5, 135.9, 133.1, 132.3, 128.4, 127.2, 125.1, 122.0, 117.7, 117.0, 115.5, 114.1; HRMS (ESI-MS) calcd for C13H11BrN2O (M + H) 291.0133, found 291.0139.:hexanes = 3:7); 1H NMR (400 MHz, CDCl3) δ 7.82 (1H, s, br), 7.59 (2H, d, J = 8.0 Hz), 7.50 (1H, d, J = 8.0 Hz), 7.39 (2H, t, J = 7.6 Hz), 7.28 (1H, t, J = 7.6 Hz), 7.17 (1H, t, J = 7.2 Hz), 6.74 (2H, d, J = 7.6 Hz), 5.51 (2H, s, br); 13C NMR (100 MHz, CDCl3) δ 167.6, 148.9, 137.8, 132.7, 129.1, 127.2, 124.5, 120.6, 117.6, 116.8, 116.3; HRMS (ESI-MS) calcd for C13H12N2O (M + H) 213.1028, found 213.1026.:hexanes = 3:7); 1H NMR (400 MHz, CDCl3) δ 11.84 (1H, s, br), 8.85 (1H, dd, J = 0.8 Hz, J = 8.8 Hz), 8.10 (1H, dd, J = 1.6 Hz, J = 8.0 Hz), 7.76 (1H, dd, J = 1.2 Hz, J = 8.0 Hz), 7.61 (1H, t, J = 7.2 Hz), 7.29 (1H, t, J = 8.4 Hz), 7.13 (1H, t, J = 8.0 Hz), 6.80 (1H, t, J = 8.0 Hz), 6.74 (1H, dd, J = 0.8 Hz, J = 8.4 Hz), 5.80 (2H, s, br), 4.00 (3H, s); 13C NMR (100 MHz, CDCl3) δ 169.0, 168.2, 149.8, 141.9, 134.6, 132.9, 131.0, 127.7, 122.3, 120.5, 117.5, 116.9, 115.7, 115.3, 52.5; HRMS (ESI-MS) calcd for C15H14N2O3 (M + Na) 293.0902, found 293.0907.:hexanes = 3:7); IR (KBr) 3296, 1632, 1612, 1506, 1326, 1263, 758 cm−1; 1H NMR (400 MHz, DMSO-d6) δ 7.68 (2H, t, J = 6.4 Hz), 7.33 (4H, t, J = 6.8 Hz), 7.28–7.24 (5H, m), 7.19 (1H, d, J = 6.8 Hz), 6.93 (2H, d, J = 8.8 Hz), 6.80 (2H, d, J = 8.0 Hz), 6.75 (2H, t, J = 7.6 Hz), 5.33 (1H, q, J = 5.6 Hz), 4.97 (1H, q, J = 5.6 Hz), 2.22 (3H, s), 2.17 (3H, s), 1.28 (3H, d, J = 5.6 Hz), 1.13 (3H, d, J = 5.6 Hz); 13C NMR (100 MHz, DMSO-d6) δ 162.5, 162.2, 148.5, 147.9, 139.43, 139.38, 137.3, 136.0, 133.91, 133.86, 131.3, 130.8, 130.6, 128.5, 128.4, 128.3, 128.2, 127.9, 127.3, 126.8, 118.0, 117.9, 115.9, 115.5, 115.3, 114.9, 67.9, 67.1, 20.5, 18.6, 17.9; m/z = 253 (M + H), positive mode. Anal. calcd for C16H16N2O: C, 76.16; H, 6.39; N, 11.10%. Found: C, 76.31; H, 6.31; N, 11.18%.:hexanes = 3:7); IR (KBr) 3057, 2915, 1687, 1605, 1468, 1342, 1282, 1068 cm−1; 1H NMR (400 MHz, CDCl3) δ 8.00 (1H, d, J = 8.0 Hz), 7.54–7.51 (1H, m), 7.41–7.31 (4H, m), 6.92 (1H, t, J = 7.6 Hz), 6.74 (1H, d, J = 8.0 Hz), 5.31 (1H, q, J = 5.2 Hz), 4.59–4.54 (1H, m), 1.38–1.34 (3H, m); 13C NMR (100 MHz, CDCl3) δ 163.2, 162.7, 146.6, 146.5, 136.8, 134.5, 133.7, 132.9, 132.3, 130.4, 130.3, 129.9, 129.3, 129.2, 127.9, 127.5, 119.6, 116.6, 115.3, 115.1, 68.4, 66.6, 20.5; m/z = 272 (M+), positive mode. Anal. calcd for C15H13ClN2O: C, 66.06; H, 4.80; N, 10.27%. Found: C, 66.15; H, 4.73; N, 10.36%.:hexanes = 3:7); IR (KBr) 3088, 2349, 1679, 1601, 1567, 1469, 1275, 757 cm−1; 1H NMR (400 MHz, CDCl3) δ 8.00 (1.2H, dd, J = 0.8 Hz, J = 7.6 Hz), 7.73–7.69 (1.2H, m), 7.43–7.31 (3.6H, m), 7.27–7.22 (1.2H, m), 6.94–6.90 (1.33H, m), 6.75–6.72 (1.29H, m), 5.32 (1.4H, q, J = 6.0 Hz), 4.62–4.57 (1.36H, m), 1.37–1.35 (3.6H, m); 13C NMR (100 MHz, CDCl3) δ 163.0, 162.9, 146.8, 146.5, 139.1, 138.5, 133.75, 133.69, 133.6, 132.4, 129.9, 129.5, 129.3, 129.27, 129.2, 128.6, 128.2, 125.1, 123.2, 119.6, 116.9, 116.6, 115.4, 115.1, 68.2, 66.7, 20.45, 20.42; HRMS (ESI-MS) calcd for C15H1379BrN2O (M + Na) 339.0109, found 339.0114; C15H1381BrN2O (M + Na) 341.0088, found 341.0099.:hexanes = 3:7); IR (KBr) 3299, 1634, 1612, 1495, 1263 cm−1; 1H NMR (400 MHz, CDCl3) δ 8.00 (1H, d, J = 8.0 Hz), 7.44 (2H, t, J = 8.0 Hz), 7.36–7.29 (4H, m), 6.90 (1H, t, J = 7.6 Hz), 6.70 (1H, d, J = 8.0 Hz), 5.23 (1H, q, J = 6.0 Hz), 4.61 (1H, s, br), 1.42 (3H, d, J = 6.0 Hz); 13C NMR (100 MHz, CDCl3 + DMSO-d6) δ 162.9, 145.9, 140.2, 133.7, 129.3, 129.1, 127.8, 127.3, 119.4, 116.7, 115.1, 68.5, 20.9; m/z = 239 (M + H), positive mode. Anal. calcd for C15H14N2O: C, 75.61; H, 5.92; N, 11.76%. Found: C, 75.52; H, 6.07; N, 11.65%.:hexanes = 3:7); IR (KBr) 3304, 2958, 2920, 1726, 1649, 1523, 1260, 958 cm−1; 1H NMR (400 MHz, CDCl3) δ 8.04 (1H, s, br), 7.98 (1H, d, J = 7.6 Hz), 7.61 (1H, t, J = 7.6 Hz), 7.45 (1H, t, J = 6.8 Hz), 7.36 (2H, d, J = 6.8 Hz), 6.93 (1H, t, J = 7.6 Hz), 6.76 (1H, d, J = 8.0 Hz), 5.41 (1H, d, J = 5.2 Hz), 4.51 (1H, s, br), 3.83 (3H, s), 1.3 (3H, s); 13C NMR (125 MHz, CDCl3) δ 166.0, 163.8, 146.5, 139.2, 134.6, 133.5, 132.9, 132.3, 132.1, 131.5, 131.0, 129.1, 128.0, 127.7, 122.4, 120.5, 119.8, 117.5, 115.7, 67.7, 52.3, 20.6; HRMS (ESI-MS) calcd for C17H16N2O3 (M + Na) 319.1059, found 319.1065.:hexanes = 3:7); IR (KBr) 3294, 1631, 1610, 1507, 1249, 1160, 747 cm−1; 1H NMR (400 MHz, CDCl3) δ 8.05 (1H, t, J = 7.2 Hz), 7.33–7.29 (5H, m), 7.22–7.19 (3H, m), 6.91 (1H, t, J = 7.2 Hz), 6.79 (2H, d, J = 7.6 Hz), 6.65 (1H, d, J = 8.0 Hz), 6.09 (1H, s), 4.77 (1H, s), 3.77 (3H, s); 13C NMR (100 MHz, CDCl3) δ 163.2, 159.9, 145.5, 140.6, 133.8, 131.9, 129.1, 128.9, 128.2, 127.1, 126.8, 119.5, 116.9, 114.8, 113.9, 74.3, 55.2; m/z = 331 (M + H), positive mode. Anal. calcd for C21H18N2O2: C, 76.34; H, 5.49; N, 8.48%. Found: C, 76.25; H, 5.41; N, 8.56%.:hexanes = 3:7); IR (KBr) 2920, 1678, 1608, 1270, 771 cm−1; 1H NMR (400 MHz, CDCl3) δ 8.18 (1H, d, J = 8.0 Hz), 7.66 (1H, t, J = 8.0 Hz), 7.59 (1H, d, J = 8.0 Hz), 7.36 (1H, t, J = 7.6 Hz), 7.30–7.23 (3H, m), 7.05 (1H, d, J = 7.2 Hz), 2.08 (3H, s), 2.02 (3H, s); 13C NMR (100 MHz, CDCl3) δ 161.0, 153.7, 147.0, 136.2, 134.7, 134.0, 130.9, 128.9, 127.3, 127.0, 126.5, 126.2, 126.0, 120.1, 23.3, 16.8; HRMS (ESI-MS) calcd for C16H14N2O (M + H) 251.1184, found 251.1181.:hexanes = 3:7); IR (KBr) 2917, 1686, 1607, 1472, 1280 cm−1; 1H NMR (400 MHz, CDCl3) δ 8.31 (1H, dd, J = 1.2 Hz, J = 8.0 Hz), 7.83–7.79 (1H, m), 7.72 (1H, d, J = 8.0 Hz), 7.66–7.62 (1H, m), 7.52–7.47 (3H, m), 7.38–7.35 (1H, m), 2.25 (3H, s); 13C NMR (100 MHz, CDCl3) δ 161.5, 153.7, 147.5, 135.5, 134.8, 132.6, 130.85, 130.81, 129.9, 128.4, 127.2, 126.9, 126.8, 120.6, 23.6; m/z = 271 (M + H), positive mode. Anal. calcd for C15H11ClN2O: C, 66.45; H, 4.10; N, 10.35%. Found: C, 66.42; H, 4.18; N, 10.26%.:hexanes = 3:7); IR (KBr) 1685, 1605, 1341, 1282, 774 cm−1; 1H NMR (400 MHz, CDCl3) δ 8.31 (2H, d, J = 7.6 Hz), 7.81 (1H, t, J = 8.0 Hz), 7.72 (1H, d, J = 7.6 Hz), 7.57–7.49 (2H, m), 7.42 (1H, t, J = 8.0 Hz), 7.38 (1H, d, J = 8.0 Hz), 2.25 (3H, s); 13C NMR (100 MHz, CDCl3) δ 161.4, 153.6, 147.6, 137.2, 134.8, 134.0, 130.9, 129.9, 129.1, 127.2, 126.9, 126.7, 122.9, 120.6, 23.7; HRMS (ESI-MS) calcd for C15H1179BrN2O (M + H) 315.0133, found 315.0128; C15H1181BrN2O (M + H) 317.0113, found 317.0109.:hexanes = 3:7); IR (KBr) 1682, 1607, 1342, 1274, 769 cm−1; 1H NMR (500 MHz, CDCl3) δ 8.28 (1H, dd, J = 1.0 Hz, J = 7.5 Hz), 7.79–7.76 (1H, m), 7.70 (1H, d, J = 8.0 Hz), 7.59–7.51 (3H, m), 7.49–7.46 (1H, m), 7.29–7.27 (2H, m), 2.26 (3H, s); 13C NMR (125 MHz, CDCl3) δ 162.3, 154.3, 147.4, 137.8, 134.6, 130.0, 129.3, 128.0, 127.1, 126.75, 126.7, 120.8, 24.4; HRMS (ESI-MS) calcd for C15H12N2O (M + Na) 259.0848, found 259.0846.:hexanes = 3:7); IR (KBr) 3052, 2964, 1600, 1419, 1260, 1095 cm−1; 1H NMR (400 MHz, DMSO-d6) δ 8.20 (1H, d, J = 8.0 Hz), 8.12 (1H, d, J = 7.6 Hz), 7.91 (2H, t, J = 8.0 Hz), 7.78–7.73 (2H, m), 7.68 (1H, d, J = 7.6 Hz), 7.57 (1H, t, J = 8.0 Hz), 3.70 (3H, s), 2.15 (3H, s); 13C NMR (100 MHz, DMSO-d6) δ 164.4, 161.3, 154.1, 147.3, 137.7, 134.6, 134.3, 131.3, 130.4, 129.7, 127.3, 126.6, 126.3, 126.2, 120.2, 52.3, 23.6; HRMS (ESI-MS) calcd for C17H14N2O3 (M + Na) 317.0902, found 317.0899.:hexanes = 3:7); IR (KBr) 1682, 1606, 1512, 1252, 774 cm−1; 1H NMR (400 MHz, CDCl3) δ 8.37 (1H, d, J = 7.6 Hz), 7.84 (2H, s), 7.56–7.54 (1H, m), 7.39–7.29 (5H, m), 7.19 (2H, d, J = 7.2 Hz), 6.74 (2H, d, J = 8.4 Hz), 3.78 (3H, s); 13C NMR (100 MHz, CDCl3) δ 162.5, 160.3, 154.9, 147.6, 137.9, 134.7, 130.8, 129.1, 129.0, 128.3, 127.8, 127.6, 127.2, 127.0, 120.8, 113.4, 55.2; m/z = 329 (M + H), positive mode; anal. calcd for C21H16N2O2: C, 76.81; H, 4.91; N, 8.53%. Found: C, 76.95; H, 5.07; N, 8.45%.Acknowledgements
We thank DST for financial support (project number: SR/S1/OC-70/2008) of the facility in our school. S. K. G also thanks UGC for senior research fellowship.Notes and references
-
(a) E. J. Corey and X. M. Cheng, in The Logic of Chemical Synthesis, Wiley, New York, 1995, pp. 1–16 Search PubMed
; (b) K. C. Nicolaou, D. Vourloumis, N. Winssinger and P. S. Baran, Angew. Chem., Int. Ed., 2000, 39, 44–122 CrossRef CAS
-
(a) N. V. Tsarevsky and K. Matyjaszewski, Chem. Rev., 2007, 107, 2270–2299 CrossRef CAS PubMed
-
(a) J. Mlynarski and J. Paradowska, Chem. Soc. Rev., 2008, 37, 1502–1511 RSC
-
(a) F. V. Rantwijk and R. A. Sheldon, Chem. Rev., 2007, 107, 2757–2785 CrossRef PubMed
-
(a) R. Noyori, Chem. Commun., 2005, 1807–1811 RSC
-
(a) D. E. Bergbreiter, J. Tian and C. Hongfa, Chem. Rev., 2009, 109, 530–582 CrossRef CAS PubMed
- M. Petkovic, K. R. Seddon, L. P. N. Rebeloa and C. S. Pereira, Chem. Soc. Rev., 2011, 40, 1383–1403 RSC
-
(a) C. Ruß and B. König, Green Chem., 2012, 14, 2969 RSC
- P. M. Pawar, K. J. Jarag and G. S. Shankarling, Green Chem., 2011, 13, 2130–2134 RSC
-
(a) G. Imperato, E. Eibler, J. Niedermaier and B. Konig, Chem. Commun., 2005, 1170–1172 RSC
- F. Ilgen and B. Konig, Green Chem., 2009, 11, 848–854 RSC
- G. Imperato, S. Hoger, D. Lenoir and B. Konig, Green Chem., 2006, 8, 1051–1055 RSC
- S. Gore, S. Baskaran and B. Konig, Green Chem., 2011, 13, 1009–1013 RSC
-
(a) S. B. Mhaske and N. P. Argade, Tetrahedron, 2006, 62, 9787–9826 CrossRef CAS
-
(a) C. A. Sutheimer, B. R. Hepler and I. Sunshine, J. Anal. Toxicol., 1983, 7, 83–85 CrossRef CAS PubMed
-
(a) C. Ma, Y. Li, S. Niu, H. Zhang, X. Liu and Y. Che, J. Nat. Prod., 2011, 74, 32–37 CrossRef CAS PubMed
-
(a) J. Zhou and J. Fang, J. Org. Chem., 2011, 76, 7730–7736 CrossRef CAS PubMed
-
(a) Y.-P. Zhu, Z. Fei, M.-C. Liu, F.-C. Jia and A.-X. Wu, Org. Lett., 2013, 15, 378–381 CrossRef CAS PubMed
- P. P. Naidu, A. Raghunadh, K. R. Rao, R. Mekala, B. J. Moses, B. R. Rao, V. Siddaiah and M. Pal, Synth. Commun., 2014, 44, 1475 CrossRef CAS
-
(a) J.-F. Liu, P. Ye, K. Sprague, K. Sargent, D. Yohannes, C. M. Baldino, C. J. Wilson and S.-C. Ng, Org. Lett., 2005, 7, 3363–3366 CrossRef CAS PubMed
- J. Chen, D. Wu, F. He, M. Liu, H. Wu, J. Ding and W. Su, Tetrahedron Lett., 2008, 49, 3814 CrossRef CAS
- P. Salehi, M. Dabiri, M. Baghbanzadeh and M. Bahramnejad, Synth. Commun., 2006, 36, 2287 CrossRef CAS
- L. M. Wang, L. Hu, J. H. Shao, J. J. Yu and L. Zhang, J. Fluorine Chem., 2008, 129, 1139 CrossRef CAS
- M. Dabiri, P. Salehi, S. Otokesh, M. Baghbanzadeh, G. Kozehgary and A. A. Mohammadi, Tetrahedron Lett., 2005, 46, 6123 CrossRef CAS
- H. R. Shaterian, A. R. Oveisi and M. Honarmand, Synth. Commun., 2010, 40, 1231 CrossRef CAS
- J. Safari and S. Gandomi-Ravandi, J. Mol. Catal. A: Chem., 2013, 371, 135 CrossRef CAS
- A. Rostami, B. Tahmasbi, H. Gholami and H. Taymorian, Chin. Chem. Lett., 2013, 24, 21 CrossRef
- W. Ge, X. Zhuab and X. Wei, RSC Adv., 2013, 3, 10817–10822 RSC
- S. B. Bharate, N. Mupparapu, S. Manda, J. B. Bharate, R. Mudududdla, R. R. Yadav and R. A. Vishwakarma, ARKIVOC, 2012, viii, 308–318 Search PubMed
- M. Rueping, A. P. Antonchick, E. Sugiono and K. Grenader, Angew. Chem., Int. Ed., 2009, 48, 908 CrossRef CAS PubMed
- A. Ghorbani-Choghamarani and T. Taghipour, Lett. Org. Chem., 2011, 8, 470 CrossRef CAS
- J. Chen, W. Su, H. Wu, M. Liub and C. Jin, Green Chem., 2007, 9, 972–975 RSC
- J. Wu, X. Du, J. Ma, Y. Zhang, Q. Shi, L. Luo, B. Song, S. Yanga and D. Hua, Green Chem., 2014, 16, 3210 RSC
- R. Sharma, A. Pandey and P. M. S. Chauhan, Synlett, 2012, 23, 2209–2214 CrossRef CAS
- B. V. S. Reddy, A. Venkateswarlu, C. Madan and A. Vinu, Tetrahedron Lett., 2011, 52, 1891–1894 CrossRef
- B. A. Dar, A. K. Sahu, P. Patidar, J. Patial, P. Sharma, M. Sharma and B. Singh, Am. J. Chem., 2012, 2, 248–254 CrossRef CAS
- M. Desroses, M. Scobie and T. Helleday, New J. Chem., 2013, 37, 3595 RSC
- Z. H. Zhang, X. N. Zhang, L. P. Mo, Y. X. Li and F. P. Ma, Green Chem., 2012, 14, 1502–1506 RSC
- H. R. Lobo, B. S. Singh and G. S. Shankarling, Catal. Commun., 2012, 27, 179–183 CrossRef CAS
-
(a) R. R. Jella and R. Nagarajan, Tetrahedron, 2013, 69, 10249 CrossRef CAS
-
(a) C. Wattanapiromsakul, P. I. Forster and P. G. Waterman, Phytochemistry, 2003, 64, 609 CrossRef CAS PubMed
- M. Viji and R. Nagarajan, J. Chem. Sci., 2014, 126, 1075–1080 CrossRef CAS
- K. Uehata, N. Kimura, K. Hasegawa, S. Arai, M. Nishida, T. Hosoe, K.-I. Kawai and A. Nishida, J. Nat. Prod., 2013, 76, 2034–2039 CrossRef CAS PubMed
-
(a) M. Sharma, K. Chauhan, R. Shivahare, P. Vishwakarma, M. K. Suthar, A. Sharma, S. Gupta, J. K. Saxena, J. Lal, P. Chandra, B. Kumar and P. M. S. Chauhan, J. Med. Chem., 2013, 56, 4374–4392 CrossRef CAS PubMed
- B. K. Oha, E. B. Koa, J. W. Hana and C. H. Oha, Synth. Commun., 2015, 45, 758–766 CrossRef
- W. M. Welch, F. E. Ewing, J. Huang, F. S. Menniti, M. J. Pagnozzi, K. Kelly, P. A. Seymour, V. Guanowsky, S. Guhan, M. R. Guinn, D. Critchett, J. Lazzaro, A. H. Ganong, K. M. DeVries, T. L. Staigers and B. L. Chenard, Bioorg. Med. Chem. Lett., 2001, 11, 177–181 CrossRef CAS PubMed
- J. Fang and J. Zhou, Org. Biomol. Chem., 2012, 10, 2389–2391 CAS
Footnote |
| † Electronic supplementary information (ESI) available: NMR, CHN, LCMS and HRMS spectra. See DOI: 10.1039/c6ra00855k |
| This journal is © The Royal Society of Chemistry 2016 |