
Preparation and dielectric properties of polymer composites incorporated with polydopamine@AgNPs core–satellite particles†
Abstract
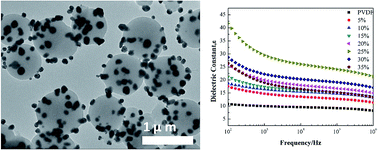
Polydopamine@AgNPs (PDA@AgNPs) core–satellite particles were fabricated by self-polymerization of dopamine and in situ reduction of Ag+ on the as-formed PDA surface. The PDA@AgNPs particles were introduced into poly(vinylidene difluoride) (PVDF) as fillers to prepare the PDA@AgNPs/PVDF composites. The employment of AgNPs could effectively increase the dielectric constant of the polymer composite. And the dielectric property of the composite could be regulated by the size of the PDA core. In addition, the core–satellite structure had the merit of preventing AgNPs from serious aggregation due to the immobilization on the PDA particles. Thanks to the bonding ability of PDA with various materials, this procedure shed light on the preparation of a number of new kinds of core–satellite particles with promising potentials in electronic fields.
 Please wait while we load your content...
Please wait while we load your content...

