
Activity and kinetics of ruthenium supported catalysts for sodium borohydride hydrolysis to hydrogen†
Abstract
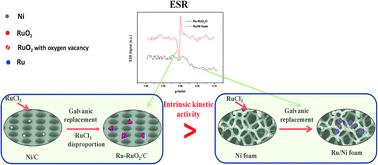
Ru–RuO2/C prepared by galvanic replacement has high catalytic activity for sodium borohydride hydrolysis. In the present study, a series of Ru–RuO2/C catalysts, Ru–RuO2/C reduced, RuO2/C and Ru supported on Ni foam (Ru/Ni foam) are prepared and characterized. Results show that RuO2 on Ru–RuO2/C is formed from both the consumption of the parent Ni and NiO nanoparticles and the disproportionation of RuCl3 with epitaxial growth of Ru species. The quantity of RuO2 with oxygen vacancies in Ru–RuO2/C determines the hydrolysis activity for sodium borohydride. In contrast to Ru–RuO2/C, Ru/Ni foam without oxygen vacancies has the lower hydrolysis activity. Results of kinetics calculation further confirm that without mass transfer limitation, Ru–RuO2/C has lower intrinsic activation energy and correspondingly higher catalytic activity due to existence of oxygen vacancies than those from Ru–RuO2/C reduced, RuO2/C, Ru/Ni foam and catalysts from the literature.
 Please wait while we load your content...
Please wait while we load your content...

