DOI:
10.1039/C6RA00727A
(Paper)
RSC Adv., 2016,
6, 25943-25951
Hemocompatible glutaminase free L-asparaginase from marine Bacillus tequilensis PV9W with anticancer potential modulating p53 expression†
Received
9th January 2016
, Accepted 29th February 2016
First published on 2nd March 2016
Abstract
Bacillus tequilensis PV9W, a marine bacterial isolate obtained from Gulf of Mannar, Rameswaram, India, produced glutaminase free L-asparaginase which was purified to homogeneity with a significant increase (13 fold) in specific activity. The apparent Km (0.045 ± 0.013 mM) and Vmax (7.465 ± 0.372 μmol ml−1 min−1) values of this purified L-asparaginase was identified and it was found that the maximum activity of the L-asparaginase was at a pH 8.5 and a temperature of 35 °C. The enzyme is a mixed α/β protein and the influence of different effectors were documented. The purified L-asparaginase had effective acrylamide degradation activity (6 IU per ml) and cytotoxic activity against HeLa cell lines with an IC50 of 0.036 ± 0.009 IU per ml. Furthermore, the purified enzyme showed p53 dependent G2 arrest in HeLa cells analyzed by FACS and was hemocompatible. Thus, this study highlights the marine isolate PV9W as a potential source for glutaminase free L-asparaginase with industrial as well as pharmaceutical applications. This study also paves a way for a possible therapeutic drug with the least amount of side effects.
Introduction
L-Asparaginase (EC 3.5.1.1) mediates the conversion of L-asparagine to L-asparatic acid and ammonia. The auxotrophic nature of some tumor cells to synthesize L-asparagine for its essential cellular processes, has highlighted the use of L-asparaginase to deplete the existing concentration of L-asparagine and thus forcing the tumor cells to undergo apoptosis.1 The therapeutic application of L-asparaginase is documented in various situations like Hodgkin disease, acute lymphoblastic leukemia, reticle sarcoma, myelocytic leukemia, lymphosarcoma, melanosarcoma and pancreatic carcinoma.2,3 This enzyme also finds an application in food industry for reduction of formation of acrylamide in fried foods.4 L-Asparaginase has been obtained and investigated from various plant and microbial sources as reviewed by many researchers.5,6 Among these the Bacillus species from diverse environments have been explored on a wide scale for its ability to survive in extreme environments, which provide stability to the enzyme derived in commercial production and purification.7–9 Further, the L-asparaginase produced from Bacillus species are regarded as safe and possess no health-concerns for use in food industry as well as in pharmaceutical applications as anticancer agents, with lesser or nil side effects.10 Moreover the glutaminase free property of L-asparaginase is essential to circumvent the problem of diminishing L-glutamine from the circulatory system, which results in various side effects in patients like seizures, hyperglycemia, leucopenia and acute pancreatitis.11 Thus, glutaminase free L-asparaginase from Bacillus sp. is an attractive perspective of research.
One such species, Bacillus tequilensis, known for its ability to survive a wide pH range, was reported to produce some commercially important enzyme like thermophilic alkalophillic proteases.12 Moreover, the gene encoding for L-asparaginase production (ansA) has been identified, cloned and reported from Bacillus tequilensis strain NIOS4.10 This study focuses on the production, purification, characterization and biological application of L-asparaginase purified from a marine isolate Bacillus tequilensis PV9W.
Materials and methods
Chemicals
Readymix Taq Polymerase Kit, Genelute kit and Sephacryl S-200 were purchased from Sigma Aldrich (USA). Ammonium sulphate was purchased from MERCK (Mumbai, India). All other laboratory chemicals and salts were purchased from Himedia, (Mumbai).
Isolation of bacterial culture and preliminary screening of L-asparaginase production
Marine isolate, (PV9W) from the sea water of the Gulf of Mannar, Ramanathapuram district, Tamil Nadu, India, was screened for L-asparaginase and L-glutaminase activity by standard flask method13 and was identified on the basis of the sequence of its amplified its 16S rDNA region (SciGenome, Kochi, India).
Quantitative estimation of L-asparaginase produced by marine isolate PV9W
L-Asparaginase was produced extracellularly, by PV9W in 50 ml of M-9 medium containing various concentration of L-asparagine (0.1–1%) as substrate and incubated at 37 °C for a period of 48 h. For the quantitative estimation of L-asparaginase, the cell free supernatant (crude enzyme) was incubated with L-asparagine as substrate for 10 min at 37 °C. The product formed, liberated ammonia was estimated by direct nesslerization.14 One unit of enzyme activity was given by that amount of enzyme which produces 1 μmol of ammonia per minute at pH 8.5 and 37 °C.14 The total protein contents of the enzyme were estimated by Lowry method where bovine serum albumin (Himedia, India) was used as a standard.
Purification and characterization of enzyme
The L-asparaginase produced extracellularly by PV9W in M-9 medium with 1% L-asparagine after incubation of 48 h at 37 °C, was purified sequentially by ammonium sulphate precipitation, dialysed against Tris–HCl buffer pH 8.5 and finally by size exclusion chromatography. All steps of purification like precipitation and dialysis were done at 4 °C to avoid loss of active enzyme.
Precipitation of L-asparaginase from crude enzyme using ammonium sulfate
The culture broth after 48 h of incubation of PV9W in M-9 medium with 1% L-asparagine was centrifuged at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 15 min. The cell free supernatant was subjected to further purification. The first step in L-asparaginase purification was by addition of powdered ammonium sulfate to the cell free extract (crude enzyme) to achieve 20% saturation and increased in succession to get the maximum precipitation of proteins (between 60–80% saturation) at the end of incubation for 4 h at 4 °C. The protein thus precipitated was suspended in 50 mM Tris–HCl buffer at pH 8.5 and subjected to dialysis (Dialysis membrane-110, Himedia, India) against the same buffer for 6 h to remove the impurities. L-Asparaginase activity present in the precipitate was estimated by direct nesslerization method. The dialyzed protein fraction was further purified by column chromatography.
000 rpm for 15 min. The cell free supernatant was subjected to further purification. The first step in L-asparaginase purification was by addition of powdered ammonium sulfate to the cell free extract (crude enzyme) to achieve 20% saturation and increased in succession to get the maximum precipitation of proteins (between 60–80% saturation) at the end of incubation for 4 h at 4 °C. The protein thus precipitated was suspended in 50 mM Tris–HCl buffer at pH 8.5 and subjected to dialysis (Dialysis membrane-110, Himedia, India) against the same buffer for 6 h to remove the impurities. L-Asparaginase activity present in the precipitate was estimated by direct nesslerization method. The dialyzed protein fraction was further purified by column chromatography.
Sephacryl S-200 gel filtration chromatography
The Sephacryl S-200 chromatography column (1 cm × 50 cm) with a bed volume of 30 ml was used for further purification. Pre-equilibration of the column was done with two bed volume of 50 mM Tris–HCl (pH 8.5), at a flow rate of 1 ml min−1. The dialyzed precipitate was loaded onto the pre equilibrated column and the adsorbed protein was eluted using the same buffer. Each fractions of 1.5 ml was eluted manually and assayed for protein at 280 nm (Biophotometer D30, Eppendorf, Germany) and L-asparaginase activity by Mashburn and Wriston (1964) method. The protein fractions were confirmed for purity and their molecular weight was identified by polyacrylamide gel electrophoresis (native and SDS treated).
Effect of physical parameters like pH, temperature and various effectors on L-asparaginase activity
The L-asparaginase purified from PV9W was characterized for its optimum pH, using different assay buffers for forming the enzyme substrate system, namely, acetate buffer (pH 4–6), potassium phosphate (pH 6.0–8.0), Tris–HCl (pH 8.0–9.5). The stability of the purified L-asparaginase was tested by incubating the purified enzyme with different buffer (pH range 4.5–9.5) for period of 24 h at 4 °C and the enzyme activity after the incubation was determined by direct nesslerization reaction and results of residual enzyme activity versus pH plotted on a graph pointed out the pH optimum for the storage of purified L-asparaginase. Similarly, the temperature at which enzyme was most active was determined by incubating the enzyme substrate system at a range of temperatures (5 °C to 55 °C) and the activity of L-asparaginase was assayed by direct nesslerization. The graph of the enzyme activity versus the incubation temperature was plotted to estimate the temperature optima.
Further, the influence of various effectors like metal ions (monovalent and divalent cations), chelating agents like ethylene diamine tetra acetic acid (EDTA), detergents like sodium dodecyl sulphate (SDS), inhibitors like phenylmethanesulfonylfluoride (PMSF) on L-asparaginase activity was estimated by incubating the purified enzyme with Na+ (NaCl – 50 mM), K+ (KCl – 150 mM), Mg2+ (MgCl2 – 40 mM), Ca2+ (CaCl2 – 150 mM), Mn2+ (MnCl2 – 100 mM, Zn2+ (ZnCl – 100 mM), Hg2+ (HgCl2 – 10 mM), Fe3+ (FeCl3 – 100 mM), EDTA (5 mM), L-cystine (25 mM), L-histidine (25 mM), mercaptoethanol (0.5 mM), SDS (2 mM) and PMSF (50 mM)) for exposure period of 30 min.11,15 The purified L-asparaginase without any effector was used as a control. The relative activity was calculated as the percentage ratio of the L-asparaginase activity when incubated with the effectors to the L-asparaginase incubated without effectors.
Secondary structure analysis using circular dichorism spectrum
The circular dichroism spectra (CD) of the L-asparaginase (4 μM) was acquired by loading into a 0.1 cm quartz cell and its ellipticity was scanned between 200 and 300 nm using a Jasco J-810 spectropolarimeter at 25 °C. The spectrum obtained is the average of three scans at scanning rate of 30 nm min−1 per sample. Further all the spectra were corrected against the base line and the graph was smoothened using Origin 8.5. Tris–HCl buffer (50 mM, pH 8.6) was used as reference and the protein was analyzed in the same buffer due to its stability at pH 8.6. Secondary structure of the purified enzyme was analyzed from the spectra of far-ultraviolet CD (far-UV CD) in the wavelength range 200–240 nm by the K2D3 software. The effect of metal ions on the secondary structure (ellipticity at 208 and 222 nm) of L-asparaginase protein (4 μM) was analyzed with 10 mM of NaCl, or MnCl2, or 200 μM of ZnCl2.
Kinetics of purified L-asparaginase of Bacillus tequilensis PV9W
The kinetics of purified L-asparaginase of Bacillus tequilensis PV9W was studied for a range of concentrations (0.01 mM to 5 mM) of its natural substrate L-asparagine. The Michaelis–Menten constant (Km) and maximal velocity (Vmax) were calculated by plotting the reaction velocity (V) reported as (μmol ml−1 min−1) vs. Substrate conc. (S) in mM by non linear regression analysis. The values of L-asparaginase activity obtained were the average of three independent analyses performed in triplicates. Graph Pad Prism Version 6 was used to analyze the data to create Michaelis–Menten plot and Lineweaver–Burk plot.
Biological applications of L-asparaginase
L-Asparaginase for the inhibition of acrylamide formation by L-asparaginase. Inhibition of polyacrylamide formation using L-asparaginase purified from Bacillus tequilensis PV9W was demonstrated.7 Briefly, to a system consisting of 10% acrylamide solution (5 ml), 2.5 ml of Tris–HCl buffer (pH 8.5) and various concentration of L-asparaginase ranging from 0.75 IU per ml to 6 IU per ml was added, and the reaction mixture was incubated at 45 °C for 30 min. After the incubation, 200 μl of ammonium persulphate and 20 μl of tetramethylethylenediamine were added to the mixture. The time taken for acrylamide solution to solidify was documented.
Application of purified L-asparaginase as a cytotoxic agent for human cervical cancer cells (HeLa cell line). The cytotoxic activity of L-asparaginase on HeLa cells lines was determined by the standard MTT assay. Briefly, HeLa cells were grown in DMEM (high glucose) (Himedia, India) media supplemented with 10% FBS and 1% penicillin/streptomycin mixture. HeLa cells were seeded on 96-well plates (1 × 104 cells per well) and incubated at 37 °C in a 5% CO2, humidified atmosphere. The cells attained 80% confluence in 12 h cells and were treated with purified L-asparaginase produced by Bacillus tequilensis PV9W with a range of concentrations (0.005 to 0.4 IU per ml) and incubated up to 36 h. The untreated HeLa cells and plain medium were kept as cell control and blank respectively. MTT was performed as per the instructions of the MTT assay kit, (Himedia, India). The absorbance corresponding to formazan crystal formation by the surviving viable cells was read at 570 nm and at 650 nm for corresponding turbidity, using 96 well plate ELISA reader. The cytotoxicity in terms of (IC50) was calculated. IC50 is that drug concentration which inhibits growth of cells by 50% relative to control, due to the treatment by L-asparaginase.
Cell cycle analysis of HeLa cells treated with purified L-asparaginase
For the study of effect of L-asparaginase treatment on the cell cycle of HeLa cells, 1 × 106 HeLa cells were seeded in 12-well plates and grown in DMEM (Himedia, India) supplemented with 10% FBS for 12 h under standard cell culture conditions. The 6 wells of confluent cells were treated with purified L-asparaginase, while 6 wells were kept un-treated (no L-asparaginase was added) as control and incubated at 37 °C in a 5% CO2, humidified atmosphere. After 24 h incubation, the cells were washed with phosphate buffer saline (PBS) (pH 7.4) (Himedia, India), trypsinized and fixed with 70% ethanol. The cells were washed and suspended in 400 μL of PBS (pH 7.4). To the above suspension 4 μl of PI (1 mg ml−1 propidium iodide) was added and incubated for 15 min in dark. The experiment was performed using flow cytometry (FACSAria III, BD Biosciences, USA). FlowJo software was used for data analysis and to discriminate the cell cycle phases.16
Study of p53 expression in HeLa cells treated with purified L-asparaginase
HeLa cells were seeded in 12-well plates (1 × 106 HeLa cells) similar to the method used for cell cycle analysis. Treatment with purified L-asparaginase (IC50 concentration) produced by Bacillus tequilensis PV9W was given for 6 wells while the remaining 6 wells containing untreated cells, were kept as control and incubated at 37 °C in a 5% CO2, humidified atmosphere for the period of 24 h. The cells were washed with PBS, trypsinized and fixed with 70% ethanol, after incubation. Further the cells were washed with BSA in PBS (5 mg ml−1) and cells were centrifuged. To the pellet, primary antibody p53 mouse monoclonal IgG1 (Santa Cruz sc-99) in a dilution of 1 in 2500, was added incubated in dark for 30 min. Then, the cells were washed with BSA/PBS solution and to the pellet the secondary antibody Alexa Fluor 488 goat anti mouse IgG (H + L) Cat no. A11001 (Invitrogen, life technologies, USA) was added (1 in 5000 dilution) and their contents were incubated for 30 min. The cells were washed with BSA/PBS and resuspended in 400 μL PBS. The samples were analyzed by flow cytometer (FACSAria III, BD Biosciences, USA). The event of expression of p53 was documented for a population of 10000 cells in each sample and expressed as percentage. This percentage of expression was compared between treated cells and control cells.
In vitro trypsin half life (t1/2)
To determine the resistance of the purified L-asparaginase to trypsin, 0.5 ml of purified asparaginase (6 IU) was added to 2.5 ml of 0.05 M phosphate buffer (pH 7.4), containing 50 IU of trypsin.17,18 The reaction mixture was vortexed vigorously, incubated at 37 °C and the L-asparaginase activity was determined at regular intervals using 100 μl of the above said mixture, for every 10 min till the enzyme was found to be active. The analysis was performed in triplicates and a graph of relative activity with respect to enzyme without trypsin treatment was plotted with respect to time and half life was calculated.
In vitro hemolysis assays
In vitro hemolysis assay was performed on human blood (erythrocytes) to investigate the effect of purified L-asparaginase, using the blood agar plate method.19 The crude and purified asparaginases (25 μl) were separately added into the wells that were previously punched on the blood agar plate. Phosphate buffer was also placed into another well as control. The presence of hemolysis was indicated as a translucent zone of clearance, when examined after incubation at 37 °C for 24 h.
Further, quantitative hemolytic assay was also performed.20 Briefly, heparinized human blood cells (erythrocytes), washed thrice with 150 mM NaCl were suspended in 100 mM sodium phosphate buffer at pH 7.4. Cells were incubated at 37 °C with different concentration (0.5, 1.5, 3 and 6 IU) of purified L-asparaginase for 24 h. The cells were centrifuged at 2500 rpm for 15 min and the optical density of the supernatant was measured at 541 nm. Sodium phosphate buffer incubated with the erythrocytes was set as blank and the blood cells incubated with distilled water were used as a positive control.
Results and discussion
Screening for L-asparaginase production in marine isolate PV9W
Marine isolate PV9W showed potential L-asparaginase production, when screened by the flask method using M-9 medium containing 0.3% L-asparagine as the only nitrogen source and phenol red indicator to identify liberated ammonia produced when the L-asparaginase hydrolyses the L-asparagine in the medium. This L-asparaginase was further found to be free of glutaminase activity by similar screening with M-9 medium containing L-glutamine as nitrogen source (ESI Fig. 1(ii)†).13 L-Asparaginase with least reactivity for L-glutamine as a substrate is of particular interest in pharmaceuticals in view to avoid the side effects produced by the depletion of L-glutamine in patients in order to clear clinical trials.21 Marine isolates with such glutaminase free L-asparaginase has been screened and reported by qualitative screening using M-9 medium with L-asparagine and L-glutamine as nitrogen sources.22 Thus the isolate PV9W which showed potential to produce L-asparaginase free of glutaminase activity was the focus of this study. Further this isolate was identified as Bacillus tequilensis by 16S rRNA sequence analysis of the amplified 16S region (ESI Fig. 1(iii)†) which exhibited 98% sequence similarity with Bacillus tequilensis 10b strain 104919.1, by BLAST analysis (http://www.ncbi.nlm.nih.gov/) (Bacillus tequilensis PV9W – GenBank Accession number: KR261609). The analysis of the phylogenetic relation of the isolate PV9W with Bacillus tequilensis 10b strain 104919.1 was done using MEGA 6 software (ESI Fig. 1(iv)†).
Moreover, the L-asparaginase produced by Bacillus tequilensis PV9W was quantified by direct nesslerization (ESI Fig. 1(v),† Table 1). The assay was performed with crude enzyme obtained by growing Bacillus tequilensis PV9W in M-9 medium containing different concentrations of L-asparagine as substrate (0.1–1% w/v). Bacillus tequilensis PV9W showed maximum enzyme activity 1.002 ± 0.020 IU per ml and specific activity of 0.833 ± 0.023 IU per mg when grown in 50 ml of M-9 medium with 1% L-asparagine after 48 h of incubation.
Table 1 Production of L-asparaginase activity by Bacillus tequilensis PV9W using minimal media M-9 with various concentration of L-asparagine as substratea
| No. |
M-9 medium + L-asparagine (% w/v) |
Enzyme units (IU per ml) |
Protein (mg ml−1) |
Specific activity (IU per mg) |
| ± indicate SD values of mean of three independent repeats in triplicates. |
| 1 |
0 |
0.021 ± 0.001 |
0.477 ± 0.115 |
0.054 ± 0.021 |
| 2 |
0.1 |
0.088 ± 0.046 |
0.609 ± 0.040 |
0.148 ± 0.079 |
| 3 |
0.2 |
0.245 ± 0.065 |
0.693 ± 0.035 |
0.355 ± 0.088 |
| 4 |
0.3 |
0.455 ± 0.018 |
0.762 ± 0.043 |
0.602 ± 0.049 |
| 5 |
0.5 |
0.582 ± 0.043 |
0.860 ± 0.060 |
0.678 ± 0.044 |
| 6 |
0.7 |
0.695 ± 0.061 |
0.899 ± 0.089 |
0.784 ± 0.096 |
| 7 |
0.8 |
0.890 ± 0.032 |
1.067 ± 0.042 |
0.835 ± 0.035 |
| 8 |
1 |
1.002 ± 0.020 |
1.202 ± 0.020 |
0.833 ± 0.023 |
Purification of L-asparaginase produced by Bacillus tequilensis PV9W
L-Asparaginase produced by Bacillus tequilensis PV9W was purified in successive steps of precipitation, dialysis and column chromatography by size exclusion. The enzyme after ammonium sulphate precipitation (between 60–80% saturation) followed by dialysis with Tris–HCl buffer pH 8.5 (for removal of ammonium ions present in excess, which interferes with assay protocol to determine liberated ammonia, thus giving false report of high enzyme activity). The enzyme was further purified to homogeneity by size exclusion chromatography (Sephacryl S 200). For each step the L-asparaginase activity and corresponding protein content was analysed (Table 2). Each of the fractions obtained by column chromatography was assayed for protein at 280 nm. The L-asparaginase activity was also estimated for each fraction by direct nesslerization. The fold increase in the purification process increased up to 13 fold by column chromatography. L-Asparaginase obtained in the fractions was verified by both native PAGE and SDS PAGE analysis for its purity (Fig. 1). The approximate size of L-asparaginase from column chromatography was assessed by native PAGE revealed a single distinct band of protein having ∼90 kDa (Fig. 1A) as compared to the crude fraction. Further, SDS PAGE analysis exhibited the size of the single subunit of the protein was ∼30 kDa, (Fig. 1B) which substantiate that this L-asparaginase may possess three subunits. Similar reports of molecular size in SDS PAGE of about 38.8 kDa, for asparaginase from Bacillus aryabhattai ITBHU02, 33.7 kDa in Bacillus licheniformis, 38 kDa in Bacillus subtilis B11-06 have been documented earlier.8,23,24
Table 2 Summary of steps in purification of L-asparaginase produced by Bacillus tequilensis PV9W, in M-9 broth containing 1% L-asparagine
| |
Vol. (ml) |
Total enzyme units (IU) |
Total protein (mg) |
Sp. activity (IU per mg) |
Yield (%) |
Fold purification |
| Crude |
500 |
401.141 |
542.857 |
0.739 |
100.000 |
|
| Amm. sulfate Ppt. dialysed (60–80%) |
52 |
90.969 |
34.914 |
2.605 |
22.677 |
3.526 |
| Column purified (Sephacryl S-200) |
13 |
78.222 |
7.676 |
10.190 |
19.500 |
13.790 |
 |
| | Fig. 1 Electrophoretic analysis of L-asparaginase produced by Bacillus tequilensis PV9W. (A) Native PAGE on 7.5% resolving gel, and (B) SDS PAGE on 12% resolving gel. M – marker, C – crude enzyme, P – purified enzyme. | |
Study of the influence of parameters like pH, temperature and metal ions on activity and stability of purified L-asparaginase
L-Asparaginase purified from Bacillus tequilensis PV9W was analyzed for the influence of different parameters namely, pH, temperature, and metal ions. The highest L-asparaginase activity of 6.50 ± 0.45 IU per ml was observed at 35 °C beyond which there was a steep decrease in the L-asparaginase activity (Fig. 2A). Similar decrease in enzyme activity beyond 40 °C was reported for L-asparaginase from Bacillus aryabhattai ITBHU02 and Bacillus subtilis B11-06, which may be due to the denaturation or modification of the active site of L-asparaginase at temperatures higher than the optimum.8,23 Further the enzyme was active between pH 7.5 to 9.5 and highest stability of the purified L-asparaginase was observed at pH 8.5 with maximum activity of 6.311 ± 0.355 IU per ml (Fig. 2B). The results are in agreement with L-asparaginase from Bacillus licheniformis RAM-8 which was active between pH 7.0 to 9.0 for a period of 24 h.24 Thus the alkaline pH favours the catalytic activity of the enzyme as well as its storage.
 |
| | Fig. 2 Characterization of L-asparaginase purified from Bacillus tequilensis PV9W. (A) Effect of temperature on L-asparaginase activity at different temperatures from 5 to 55 °C after 30 min incubation. (B) Effect of pH of assay buffer at 37 °C on L-asparaginase activity and the stability of L-asparaginase when stored at 4 °C for 24 h at different pH 3.5 to 9.5. (C) Influence of various effectors on the activity of L-asparaginase purified from Bacillus tequilensis PV9W. (D) Enzyme kinetics given by Michaelis–Menten plot of reaction velocity (V) i.e. enzyme activity per min per ml of the enzyme vs. substrate conc. (S) in mM. Parameters Vmax 7.46 μmol ml−1 min−1 and Km 0.045 mM were calculated by non-linear regression analysis. (E) Enzyme kinetics depicted by corresponding Lineweaver–Burk plot. Error bars in all the above graphs (A–E) represent SD of three independent repeats in triplicates (F) CD spectra of purified L-asparaginase. (G) Effect of metal ions Mn2+, Zn2+ and Na+ on L-asparaginase. Shown are the mean residue ellipticities of CD spectra for L-asparaginase (solid continuous line) and in presence of different monovalent and divalent cations such as NaCl (dotted line), MnCl2 (dashed dotted line) and ZnCl2 (dashed line). Changes in secondary structure were monitored by scanning from 200 to 260 nm. | |
The influence of various effectors like metal ions like Na+, K+, Mg2+, Ca2+, Mn2+, Zn2+, Hg2+, Fe3+, metal chelator, (EDTA), amino acids like L-cystine and L-histidine, mercaptoethanol, detergent SDS and inhibitor PMSF was analyzed for its influence on L-asparaginase activity. The relative activities depicted as percentage for the enzyme exposed to these effectors with respect to the unexposed, is as given in Fig. 2C. The detrimental effect of di valent cations like Hg2+, Ca2+and Zn2+ was similar to earlier reports of L-asparaginases from Bacillus sp.8,24 This effect may be because of the presence of sulfhydryl groups in the enzyme–substrate complex which may decrease the enzyme activity.15 L-Asparaginase retained nearly 90% activity in the presence of PMSF which is a serine protease inhibitor, and hence it obviously indicates that it does not belong to serine hydrolase. Though, the same observation was documented for L-asparaginase for Bacillus sp. by Mahajan et al., (2014), it is of interest that, the L-asparaginase purified from Bacillus tequilensis PV9W retained more residual activity than Bacillus sp. reported by Mahajan et al.24 The nature of the influence of the effectors helps determine the amino acids present in the active site of the enzymes and the nature of the inhibition pattern defines the group of the enzyme under study. In this study the L-asparaginase retained more than 50% of its activity for most of the effectors tested with, except mercaptoethanol and divalent cations (highlighting presence of sulfhydryl groups in active site of the enzyme). Hence the robust nature of the enzyme is of particular interest in medical applications.
Kinetic parameters of L-asparaginase
The affinity between the L-asparaginase and its substrate, L-asparagine was studied for a range of concentrations (0.01 mM to 5 mM) of the substrate. The non-linear regression analyses of the same was done by plotting the reaction velocities (V) (enzyme activity per minute per ml – μmol ml−1 min−1) vs. substrate conc. (S) (mM) in a Michaelis–Menten plot. The Vmax and Km of L-asparaginase purified from Bacillus tequilensis PV9W was 7.46 μmol ml−1 min−1 and 0.045 mM respectively (Fig. 2D). The corresponding Lineweaver–Burk plot is depicted by Fig. 2E. The Km values tend to be different for L-asparaginases from different microbial origin and values as low as 1.4 × 10−5 M was reported from Bacillus licheniformis24 and some higher value of 0.257 mM and 0.43 mM were reported for Bacillus aryabhattai ITBHU02 (ref. 8) and Bacillus subtilis B11-06.23 Lower Km values of substrate are indication of better affinity of the enzyme to its substrate. L-Asparaginase produced by Bacillus tequilensis PV9W in this study shows noticeably low Km which can be an advantage when prospected for in vivo use as a therapeutic, where the natural availability of the substrate L-asparagine would definitely be low.
Circular dichroism and secondary structure analysis of L-asparaginase
The circular dichroism spectra (CD) spectrum of L-asparaginase purified from Bacillus tequilensis PV9W revealed negative peaks at 208 and 222 nm which indicate the presence of α helix and negative peak between 215 nm and 220 nm indicating β sheet (Fig. 2F). The K2D3 analysis of the CD spectrum in the far UV range from 200 to 240 nm predicted alpha helix to be 43% and β sheet: 6.59%. The presence of higher alpha helix to low β sheet percentages has been documented earlier in Bacillus sp.24 Further, the effect of metal ions on the secondary structure of the enzyme was analyzed using buffer containing different cations such as 10 mM NaCl, or 10 mM MnCl2, or 200 μM of ZnCl2. The recorded spectra did not show any significant changes in the ellipticity suggesting that metal ions do not cause any changes in the secondary structure at these concentrations (Fig. 2G). Thus the CD spectrum of the native L-asparaginase from Bacillus tequilensis PV9W revealed that the enzyme exhibits predominant helical structure.
Biological application of L-asparaginase
Effect of L-asparaginase on polyacrylamide formation. Acrylamide is known to be a potential carcinogen, as defined by International Agency for Research on Cancer (IARC) in 1994. Its neurotoxic and genotoxic nature to both somatic cells and germ cells were confirmed by European Commission Scientific Committee on Food (ECSCF) in 2002. Reports of its carcinogenic nature to ovarian cells and renal cells were documented by Hogervorst et al., (2007).25 Moreover risks of developing breast cancer due to the exposure to acrylamide were reported by Olesen et al., (2008).26 Acrylamide is formed as a result of reaction of L-asparagine and hexose starches in starch containing food in high temperatures prevalent in food processing steps. In this study, the L-asparaginase purified from Bacillus tequilensis PV9W was capable of hydrolyzing the free L-asparagine in a concentration dependent manner, as confirmed by the time taken to solidify for the polyacrylamide solution (Table 3). It was already documented that L-asparaginase purified from Bacillus licheniformis had shown inhibition of acrylamide formation.24 Thus this study suggests that L-asparaginase purified from Bacillus tequilensis PV9W to be capable of commercial application in food industries.
Table 3 Time required for polymerization of acrylamide in the presence of various concentrations of L-asparaginase
| No. |
Acrylamide solution (10% w/v) (ml) |
Enzyme units (IU per ml) |
Time taken for solidification (min) |
| 1 |
5 |
0 |
3.270 ± 0.880 |
| 2 |
5 |
0.75 |
5.497 ± 1.237 |
| 3 |
5 |
1.5 |
11.451 ± 3.941 |
| 4 |
5 |
3 |
23.182 ± 9.452 |
| 5 |
5 |
6 |
30.690 ± 7.890 |
Effect of purified L-asparaginase as a cytotoxic agent for human cervical cancer cells (HeLa cell line)
In vitro cytotoxicity study (MTT assay). HeLa cells were used to assess the cytotoxic activity of L-asparaginase purified from Bacillus tequilensis PV9W. The viability of HeLa cells were tested by MTT assay, when the cells were subjected to various concentration of L-asparaginase for 24 h. The results were documented as percentage viability vs. concentration of L-asparaginase used in Fig. 3 A. The IC50 value of L-asparaginase to HeLa cells was calculated (0.036 ± 0.009 IU per ml). L-Asparaginases from different sources have demonstrated different IC50 for different cell lines. However, L-asparaginase purified enzyme from Salinicoccus sp. MKJ997975 showed IC50 value of 0.171 IU per ml against HeLa.27 Thus, L-asparaginase from Bacillus tequilensis PV9W showed effective cytotoxicity on HeLa cells with relatively low IC50 value, and further the HeLa cells were analyzed for the effect of the L-asparaginase on its cell cycle regulation.
 |
| | Fig. 3 (A) Cytotoxic effect of purified L-asparaginase on HeLa cell lines. The cells were treated in triplicates with each concentration for 24 h and the cell viability was determined by the MTT assay. Error bars indicate SD of mean of three independent repeats in triplicates (n = 9). (B) The effect of purified L-asparaginase on the cell cycle analysis in HeLa cell line using flow cytometry [i] control, [ii] L-asparaginase treated. | |
Cell cycle analysis. The cell cycle analysis gives an insight of the stage at which a drug is capable of inducing an arrest. Thus, to know the exact stage of cell cycle arrest, the HeLa cells were subjected to IC50 concentration of L-asparaginase for 24 h and fixed using ethanol. The L-asparaginase treated (IC50 = 0.036 ± 0.009 IU per ml) cells showed higher percentage of G2 phase (32%) compared to untreated control cells (3%) (Fig. 3B). The mechanism of L-asparaginase to induce cell death depends on the auxotrophic nature for the amino acid L-asparagine in the cells that they are tested in.28 L-Asparagine deprivation the Jensen sarcoma has reported inhibition in the G1, S, and G2 phases of the cell cycle, however the same was not found in M phase as it was suggested that L-asparagine was required only until the initiation of mitosis. The delay and the inhibition of G2 cells from entering into mitosis is similar to that observed in the presence of the inhibitor of protein synthesis, puromycin.29 Reports of G2 to M phase arrest caused by L-asparaginase have been documented in Hep-2 cells (human epidermoid larynx carcinoma) which was attributed to the decrease in the expression of key regulatory proteins that govern G2/M transition, that include cyclin B1, Cdc 25B and Cdc 25C.30 L-Asparaginase mechanism of arrest in cell cycle has also been referred to as transient arrest which may appear at early time points (within 24 h) of enzyme treatment.18
Effect of purified L-asparaginase on p53 expression in HeLa cells
The p53 tumor suppressor protein plays a critical role in the DNA damage-induced signaling pathway and decides the outcome of DNA damage at the cellular level. HeLa cells possess integrated human papilloma virus (HPV) – 18, hence they naturally expresses the E6 onco-protein, which causes degradation of p53 protein in the ubiquitin mediated manner and thus HeLa cells are p53 deficient.31 However, there is a low basal p53 activity as seen when measured with some reporter assays, in the cervical cancer cells which are left un-treated.32 In the present study, the L-asparaginase treated HeLa cells were investigated for p53 expression by FACS analysis and it was observed that the p53 expression in L-asparaginase treated cells were higher (32.1%) than the untreated control (2.5%) (Fig. 4). Thus the conclusion of the cell cycle analysis and the p53 expression in HeLa cells treated with L-asparaginase, shows G2 arrest in cervical cancer cells in a p53 dependent pathway. G2 arrest was attributed to deprivation of L-asparagine, an essential amino acid for protein synthesis. The means by which p53 retards the cells at the G2 stage, is by inhibiting the Cdc2 through the cyclin-dependent kinase. Thus the Cdc2 does not bind Cyclin B1 and thus repression of cyclin B1 further blocks the transition of the cells into mitosis as reviewed by Taylor and Stark (2001).33
 |
| | Fig. 4 Effect of L-asparaginase on p53 expression in HeLa cells by FACS analysis. (A). Depicted as histogram. Statistics of event of p53 expression in cells. (B) Control untreated cells (2.5% p53 expression). (C) Cells treated with L-asparaginase at IC 50 value. L-Asparaginase treated HeLa cells showed 32% p53 expression compared to the untreated control. | |
In vitro tests for trypsin half life and hemocompatibility of L-asparaginase
Trypsin is present as a major component in human circulatory system that is capable of degrading or inactivating the L-asparaginase administered as a drug thus decreasing its availability in vivo. Thus L-asparaginase with a higher half-life is a matter of interest. In this study, L-asparaginase purified from Bacillus tequilensis PV9W showed a higher half life of 42.59 min (Fig. 5A) which was comparable to previous report by Husain et al., (2015).18 Higher half life generally lowers the frequency of the administration of the drug, thus reducing possibilities of side effects.17
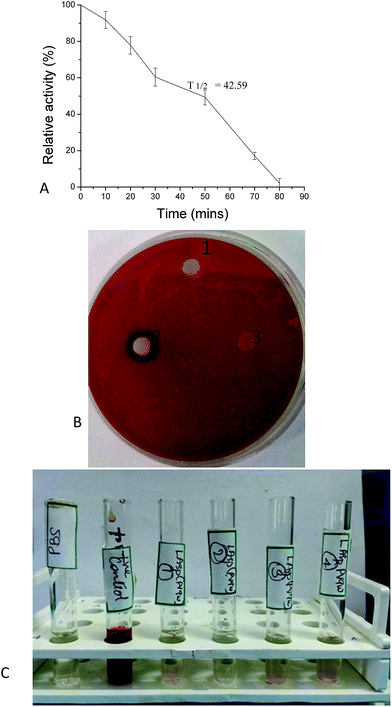 |
| | Fig. 5 Physiological properties of L-asparaginase purified from Bacillus tequilensis PV9W. (A) In vitro half-life of L-asparaginase in trypsin. Error bars indicate SD of mean of triplicate (B) Hemolytic effect of purified and crude asparaginase on human erythrocytes. (1) PBS (control), (2) crude L-asparaginase from Bacillus tequilensis PV9W, (3) purified L-asparaginase from Bacillus tequilensis PV9W. (C) Quantitative measurement of the hemolytic activity. PBS: blank (sodium phosphate buffer); +ve control: distilled water, tubes 1, 2, 3, and 4 have 0.5, 1.5, 3, and 6 IU of purified L-asparaginase. | |
Moreover, the toxicity of the L-asparaginase to erythrocytes was estimated qualitatively and quantitatively (Fig. 5B and C) and the pure enzyme was found to have no toxic effect on the erythrocytes even up to 6 IU. Similar effects have been documented for L-asparaginase from Pseudomonas otitidis and Bacillus licheniformis.18,24 Hemolysis has been reported to be the major drawbacks of many drugs used in therapeutics for which, a wide number of side-effects of toxicity to blood cells are reported.34 Thus the L-asparaginase purified from Bacillus tequilensis PV9W which was observed to be hemocompatible is definitely a matter of interest in developing field of pharmaceuticals.
Conclusion
L-Asparaginase produced by Bacillus tequilensis PV9W was purified to homogeneity, and it was a homo-trimer having molecular size of about 90 kDa. L-Asparaginase was characterized for its optimum temperature; pH and its kinetics were studied. The analysis of its secondary structure by CD spectra resulted into an α + β mixed protein. The application of the enzyme in acrylamide degradation and its cytotoxicity in HeLa cells was studied. This study highlights a p53 mediated G2 arrest of cell cycle in L-asparaginase treated HeLa cells. Moreover the higher half-life in trypsin and its hemocompatibility makes it a potential candidate for an anti cancer drug.
Acknowledgements
Authors thank the DST-SERB-SB/EMEQ-128/2013 date of sanction 28.10.2013, for the financial support to PV and CSIR-UGC (no. 17-06/2012(i) EU-V) fellowship to GS. Also the help of DST- PURSE instrumentation facility, DST-FIST, School of Biological Sciences, Madurai Kamaraj University for FACS facility and Indian Institute of Technology, Kanpur for CD spectroscopy are acknowledged.
Notes and references
- J. D. Broome, J. Exp. Med., 1968, 127, 1055 CrossRef CAS PubMed.
- A. A. Abbas, M. A. Sabbah and O. A. Kathum, Iraqi J. Sci., 2010, 51, 290 Search PubMed.
- S. Biagiotti, M. F. Paoletti, A. Fraternale, L. Rossi and M. Magnani, IUBMB Life, 2011, 63, 621 CrossRef CAS PubMed.
- E. Tareke, P. Rydberg, P. Karlsson, S. Eriksson and M. Tornqvist, J. Agric. Food Chem., 2002, 50, 4998 CrossRef CAS PubMed.
- T. Batool, E. A. Makky, M. Jalal and M. M. Yusoff, Appl. Biochem. Biotechnol., 2015 DOI:10.1007/s12010-015-1917-3.
- S. Zuo, T. Zhang, B. Jiang and W. Mu, Appl. Microbiol. Biotechnol., 2015, 99, 1069 CrossRef CAS PubMed.
- R. V. Mahajan, S. Saran, K. Kameswaran, V. Kumar and R. K. Saxena, Bioresour. Technol., 2012, 125, 11 CrossRef CAS PubMed.
- Y. Singh and S. K. Srivastav, Indian J. Exp. Biol., 2013, 51, 322 CAS.
- B. Pradhan, S. K. Dash and S. Sahoo, Asian Pac. J. Trop. Biomed., 2013, 3, 936 CrossRef CAS PubMed.
- S. Nayak, S. Porob, A. Fernandes, R. M. Meena and N. Ramaiah, J. Basic Microbiol., 2014, 54, 162 CrossRef CAS PubMed.
- S. Manna, A. Sinha, R. Sadhukhan and S. L. Chakrabarty, Curr. Microbiol., 1995, 30, 291 CrossRef CAS PubMed.
- I. Khan, P. Gupta and J. Vakhlu, Afr. J. Microbiol. Res., 2011, 5, 3968 CAS.
- R. Gulati, R. K. Saxena and R. Gupta, Lett. Appl. Microbiol., 1997, 23, 6 Search PubMed.
- L. T. Mashburn and J. C. Wriston, Arch. Biochem. Biophys., 1964, 105, 450 CrossRef CAS PubMed.
- S. C. Warangkar and C. N. Khobragade, Enzyme Res., 2010, 165878, 1 CrossRef PubMed.
- S. S. D. Kumar, A. Mahesh, S. Mahadevan and A. B. Mandal, Biochim. Biophys. Acta, 2014, 1840, 1913 CrossRef CAS PubMed.
- M. R. Tabandeh and M. Aminlari, J. Biotechnol., 2009, 141, 189 CrossRef CAS PubMed.
- I. Husain, A. Sharma, S. Kumar and F. Malik, Biochimie, 2015, 121, 38 CrossRef PubMed.
- S. Huang, G. Pan, S. Chen, Z. Huang, Y. Zhang and Z. Liang, Int. J. Biol. Macromol., 2013, 62, 124 CrossRef CAS PubMed.
- V. Bulmus, M. Woodward, L. Lin, N. Murthy, P. Stayton and A. Hoffman, J. Controlled Release, 2003, 93, 105 CrossRef CAS PubMed.
- U. K. Narta, S. S. Kanwar and W. Azmi, Crit. Rev. Oncol. Hematol., 2007, 61, 208 CrossRef PubMed.
- G. Shakambari, B. M. Sumi, B. Ashokkumar, P. Palanivelu and P. Varalakshmi, RSC Adv., 2015, 5, 48729 RSC.
- M. Jia, M. Xu, B. He and Z. Rao, J. Agric. Food Chem., 2013, 61, 9428 CrossRef CAS PubMed.
- R. V. Mahajan, V. Kumar, V. Rajendran, S. Saran, P. C. Ghosh and R. K. Saxena, PLoS One, 2014, 9, e99037 Search PubMed.
- J. G. Hogervorst, L. J. Schouten and E. J. Konings, Cancer Epidemiol., Biomarkers Prev., 2007, 16, 2304 CAS.
- P. T. Olesen, A. Olsen and H. Frandsen, Int. J. Cancer, 2008, 122, 2094 CrossRef CAS PubMed.
- M. Bhat and T. Marar, Int. J. Curr. Microbiol. Appl. Sci., 2015, 4, 701 Search PubMed.
- C. Scotti, P. Sommi, M. V. Pasquetto, D. Cappelletti, S. Stivala, P. Mignosi, M. Savio, L. R. Chiarelli, G. Valentini, V. M. Bolanos-Garcia, D. S. Merrell, S. Franchini, M. L. Verona, C. Bolis, E. Solcia, R. Manca, D. Franciotta, A. Casasco, P. Filipazzi, E. Zardini and V. Vannini, PLoS One, 2010, 5, e13892 Search PubMed.
- M. K. Patterson Jr and M. D. Maxwell, Cancer Res., 1970, 30, 1064 CAS.
- H. H. Hassani and R. J. Toma, Journal of Biotechnology Research Center, 2010, 4, 46 Search PubMed.
- B. A. Werness, E. M. Livanos and P. M. Howley, Science, 1990, 248, 76 CAS.
- S. Hietanen, S. Lain, E. Krausz, C. Blattner and D. P. Lane, Proc. Natl. Acad. Sci. U. S. A., 2000, 7, 8501 CrossRef.
- W. R. Taylor and G. R. Stark, Oncogene, 2001, 20, 1803 CrossRef CAS PubMed.
- M. M. Lubran, Ann. Clin. Lab. Sci., 1989, 19, 114–121 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra00727a |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 




