
Structure–activity relationship and binding mode studies for a series of diketo-acids as HIV integrase inhibitors by 3D-QSAR, molecular docking and molecular dynamics simulations†
Abstract
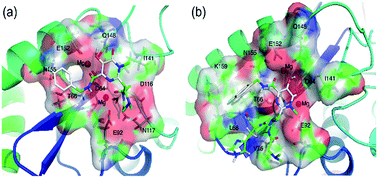
At present, the approved HIV integrase (IN) inhibitors are all diketo-acids (DKAs). To have a better understanding of DKA inhibitors, comparative molecular field analysis (CoMFA), comparative molecular similarity indices analysis (CoMSIA), docking, and molecular dynamics (MD) were performed on 88 DKAs. The constructed CoMFA and CoMSIA models were shown to be statistically significant for the training set with the cross-validated value (q2) of 0.94, non cross-validated value (r2) of 0.96 for CoMFA; q2 of 0.96, r2 of 0.94 for CoMSIA. External q2 of the test set Q2F1, Q2F2 and Q2F3 were 0.79, 0.77 and 0.81 for CoMFA; Q2F1, Q2F2 and Q2F3 were 0.60, 0.56 and 0.63 for CoMSIA. Further interpretation of contour maps provided instructive insights into the optimization and designing of lead compounds. The interaction between IN and two DKAs (the inhibitors with the highest and lowest activity) were investigated using ‘relaxed receptor’ molecular docking and molecular dynamics simulations. Molecular mechanics Poisson–Boltzmann surface area (MM-PBSA) and molecular mechanics Generalized-Born surface area (MM-GBSA) were employed to evaluate the binding affinity and the energy decomposition for residues, respectively. Further analysis indicated that the dominating effect of van der Waals drives the binding of IN and DKA inhibitors and key residues. In addition, we found that the MD conclusion was consistent with QSAR models after the analysis of the interaction between DKAs and each residue. These findings may be of immense importance in the development of DKA inhibitors.
 Please wait while we load your content...
Please wait while we load your content...

