
Rational design of benzodithiophene based conjugated polymers for better solar cell performance
Ranjith Krishna Pai
*a,
Ahipa T. N.
b and
Hemavathi B.
b
aTechnology Mission Division, Department of Science and Technology (DST), Ministry of Science and Technology, Government of India, Technology Bhavan, New Mehrauli Road, New Delhi-110016, India. E-mail: ranjith.krishnapai@gov.in; ranjith.krishnapai@gmail.com
bCNMS, Jain University, Jain Global Campus, Bangalore, India
First published on 18th February 2016
Abstract
The rational design of conjugated polymers is crucial. As photovoltaic materials they need to be optimized for high-performance polymer solar cells (PSCs). We present a concise review of conjugated polymers based on benzodithiophenes (BDTs). In this account, we have discussed the conjugated polymeric designs of various architectures; consisting of electron rich donor and electron deficient acceptor moieties in the main chain, as well as in the side chains, to facilitate effective intra-molecular charge transfer. We summarize that the application of these polymeric materials drastically influences the power conversion efficiency (PCE) of polymeric solar cells. As of now, PCEs of over 10% are reported for these polymeric materials along with fullerene derivatives.
 Ranjith Krishna Pai | Dr Ranjith Krishna Pai received a Ph.D. degree in Natural Sciences, from Dr Othmar Marti’s group, Ulm University, Germany, in 2005. He is a Senior Scientist at the Ministry of Science and Technology, Department of Science and Technology, Government of India, New Delhi, India. From 2006 to 2007, he was a Postdoctoral Researcher at the University of Chile, Santiago. From 2007 to 2009, he spent two years as a Post-Doctoral Scientist at Stockholm University, Stockholm, Sweden. From 2009 to 2011, he was a Research Scientist at CFN, Brookhaven National Laboratory, New York, USA. Dr Pai spent another 2 years 4 months (2011–2013) as a Research Scientist at INL, Braga, Portugal. From 2013 to 2015, he was an Associate Professor and Group Leader at Jain University, Bangalore, India. His research interests include energy conversion technologies, including low cost photovoltaics (organic and hybrid solar cells), electrical energy storage (batteries and supercapacitors), synthesis of semiconducting polymers and polymer nanostructures and their application to organic transistors, solar cells, light emitting diodes, and other photonic applications, syntheses, characterizations and applications of carbon and inorganic nanotubes, and modelling of the electronic properties of nanostructured semiconductors. He has been awarded the outstanding scientific research output award (Science and Technology) from the Vice Chancellor of the Jain University, Bangalore on 29th September 2015. On 19th December 2015, he was selected for the “Outstanding Scientist Award” in the category of Thin Film Solar Cells (Science) by the Venus International Foundation, Chennai, India. |
 Ahipa T. N. | Dr Ahipa T. N. obtained his five year Integrated M.Sc. (Hons) degree (2009) in Applied Chemistry at the Sahyadri Science College, Shimoga, India. Later, he obtained his Ph.D. degree (2014) in chemistry at the National Institute of Technology, Karnataka, under the direction of Prof. Airody Vasudeva Adhikari. From 2014 onwards, he was a Senior Research Associate working with Dr Ranjith Krishna Pai’s research group and projects. Recently, he was promoted as an Assistant Professor at Jain University, Bangalore, India. His research involved luminescent liquid crystals, conjugated polymers, polymer solar cells, and photoluminescent materials. |
 Hemavathi B. | Hemavathi B. obtained her BE degree in 2003 in Polymer Science and Technology from Sri Jayachamarajendra College of Engineering, Vishweshwaraiah Technological University, Mysore, India and received an M.Sc. in chemistry from Kuvempu University, India in 2011. Recently she completed her M.Phil in chemistry at Jain University, Bangalore under the guidance of Dr Ranjith Krishna Pai. Her research interests include the design and synthesis of organic/polymer materials for photovoltaic cells. |
Introduction
In recent years, considerable progress has been made on the development of conjugated polymers for photovoltaic applications. Subsequently, polymer solar cells (PSCs) with a bulk heterojunction (BHJ) structure have been researched extensively.1,2 To date, BHJ solar cells, with an active layer comprising an interpenetrating network of p-type conjugated polymers and n-type fullerene derivatives, have been comprehensively studied and have displayed the highest efficiencies of over 10%. A widely used polymer as a donor material in PSCs is poly(3-hexylthiophene) (P3HT),3–11 which has produced power conversion efficiencies (PCEs) of up to ∼5% when combined with a soluble fullerene derivative, (6,6)-phenyl C61-butyric acid methyl ester (PCBM), as the acceptor.7,12 However, further improvement of the efficiency of P3HT-based PSCs is found to be difficult because of its intrinsic limited absorption in the solar spectrum (energy band gap of 1.9 eV) and the relatively small energy difference between its highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) of PCBM. Hence, there is a quest for new structural entity based polymers to obtain high performance polymer solar cells and it is imperative to design new electron-donating polymer materials that possess a strong and broad absorption to harvest more sunlight, a high charge mobility, and low-lying HOMO energy levels, while keeping the band gap between 1.2 and 1.9 eV.13–15 The aforementioned criteria can be fulfilled only when employing a donor–acceptor (D–A) approach with alternating electron-rich and electron-deficient moieties along the polymeric backbone.13,16–19 Also, the band gaps of these polymers can be easily tuned by controlling intramolecular charge transfer (ICT) between the donor and acceptor moieties. Interestingly, a common feature of many of these efficient low band gap donor–acceptor type conjugated polymers in PSCs is the employment of the benzo[1,2-b:4,5-b′]dithiophene (BDT) unit. The polymers based on BDT showed exceptional performance as a common unit in PSCs that can achieve PCEs of up to 10%.20–22In general, a BDT unit is a planar conjugated molecule that provides for easier π–π stacking and possesses a relatively high hole mobility, which further makes the BDT based polymer able to achieve impressive photovoltaic properties for PSC applications. In this context, the present review article is mainly focused on the exploration of various polymers encompassing BDT units and their photovoltaic performances.
The presence of an alkoxy group on BDT makes BDT a very good donor and helps to increase the HOMO level to −5.16 eV.23 Homopolymers of BDT with dialkoxy and dithioalkoxy side chains were synthesised by Ferraris et al. The obtained polymers P1, P2 and P3 had a lower HOMO compared to P3HT homopolymers and polymer P3, with a dithioalkoxy side chain, exhibited an efficiency of 4.0% for the PSC (Fig. 1).24 In these polymers, the replacement of the alkoxy groups with thioalkoxy groups lowered the HOMO energy level of the conjugated polymers, and consequently enhanced the Voc. A Voc of 0.83, 0.99, and 0.91 V was obtained for polymers P1, P2 and P3, respectively. The highest Voc in polymer P2 can be attributed to the poorer electron-donating properties of the thioalkoxy group in it compared to the alkoxy group in polymer P1, which led to the decreased HOMO and LUMO energy levels of the polymers.
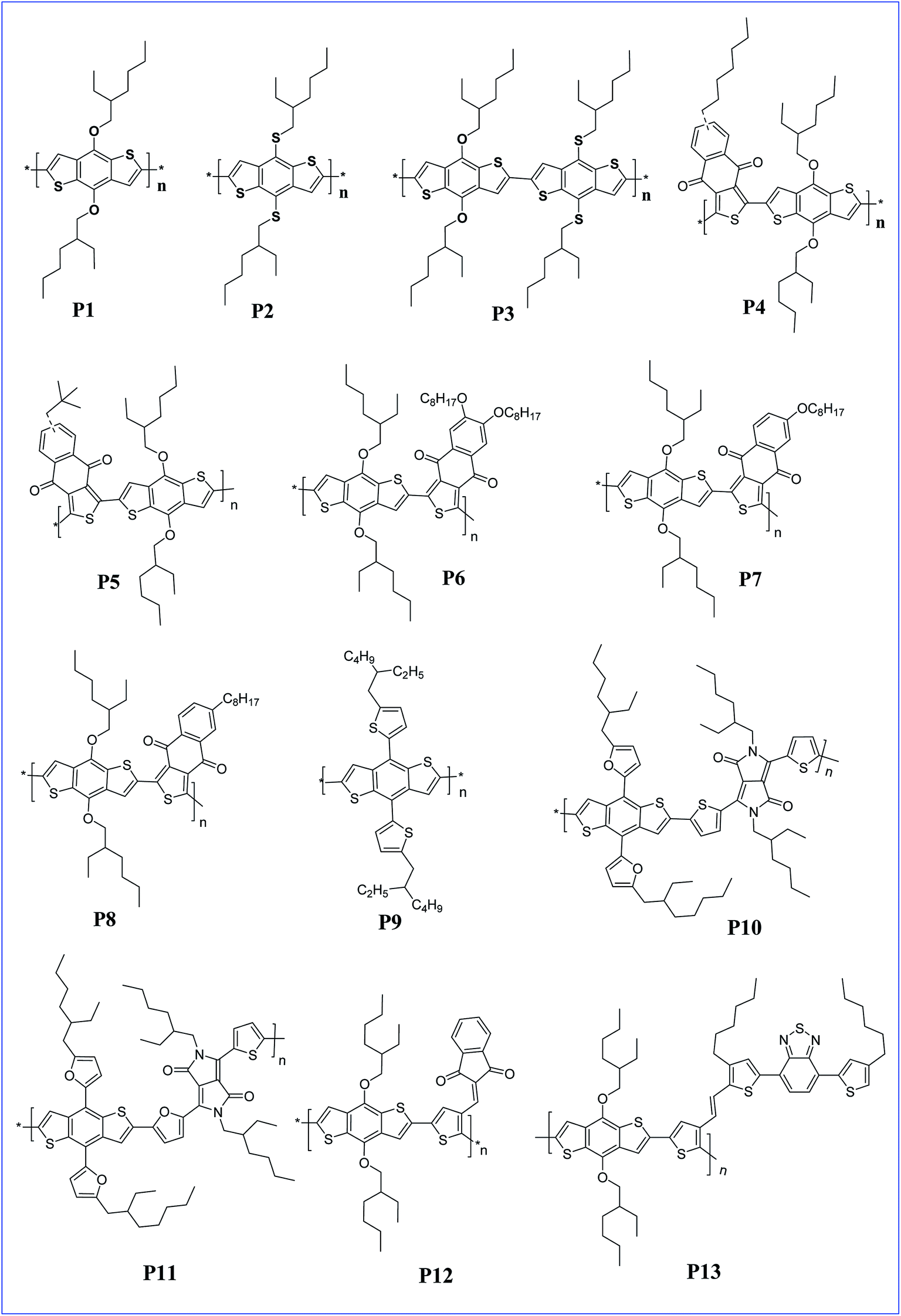 | ||
| Fig. 1 BDT based polymers with dialkoxy and NTDO derivatives in the side chains (P1–P13). | ||
D–A alternative copolymers of dialkoxy BDT with naphtho[2,3-c]thiophene-4,9-dione (NTDO) with different alkyl side chains were synthesised to afford polymers P4 and P5. The two polymers exhibit good solubility in common organic solvents due to the presence of alkyl side chains, broad visible absorption from 350 nm to 670 nm and relatively deeper HOMO energy levels (−5.14 eV for P4 and −5.19 eV for P5). Due to the strong electron withdrawing property of NTDO, both the HOMO and LUMO energy levels decreased and lead to a Voc of 0.81 and 0.88 V for P4 and P5, respectively.25 These results indicate that the short and branched alkyl chains on NTDO in P5 are more beneficial for photovoltaic applications. Similarly, Zou and co-workers synthesised a few more copolymers from the dialkoxy BDT and NTDO derivatives bearing different side chains like octyl and octyloxy groups at different positions on NTDO to afford polymers P6, P7 and P8. These polymers also showed good solubility in common organic solvents and a broad absorption from 300–700 nm. Further, the device performance is better for P8 containing an octyl side group compared to the octyloxy side chain on NTDO derivatives.26
Recently, Kim and co-workers reported a homopolymer of poly(4,8-bis(5-(2-ethylhexyl)thiophen-2-yl)benzo[1,2-b:4,5-b′]dithiophene) P9 which has high light absorption coefficients and nearly perfect energy alignment with that of [6,6]-phenyl-C71-butyric acid methyl ester (PC71BM).27 The device constructed with the P9 and PC71BM combination was able to produce high-performance PSCs with a power conversion efficiency (PCE) of 6.12% and a high Voc (0.93 V). The obtained PCE value is one of the highest values reported for homopolymer donor-based PSCs and is found to be a superior polymer compared to P3HT in terms of wide band gap polymers. The presence of conjugated side chains on the polymer main chain facilitated efficient hole mobility and broadens the absorption range, thus, enhanced the PSCs.28
The effects of alkylfuranyl-substituted conjugated donor–acceptors based on BDT polymers were explored by Ge and colleagues. Here, furanyl group introduced polymers, P10 and P11 exhibited a reduced band gap and increased absorption range, as well as influencing the performance of PSCs.29 The efficiency of PSCs based on polymers P10 and P11 reached 3.5% and 5.1%, respectively, whereas PSCs based on the replacement of alkylfuranyl with alkoxy substituents in these polymers showed an efficiency of only 1.0% and 2.9%. Further, copolymers (P12 and P13) of BDT and thiophene with different conjugated side groups like 4,7-dithien-5-yl-2,1,3-benzodiathiazole (DTBT) as electron-deficient units were synthesized by Tan and co-workers. The introduced side chain conjugation lowers the HOMO level, and the bulk heterojunction solar cell prepared by these polymers with PC61BM exhibited a PCE of 2.48% and 4.18% for P12 and P13, respectively.30 In addition, three copolymers (P14, P15 and P16) of BDT and thiophene with different conjugated side chains introduced, such as di(p-tolyl)phenylamine (TPA), 4,7-dithien-5-yl-2,1,3-benzothiadiazole (DTBT) and 4,7-dithien-5-yl-2,1,3-benzothiadiazole–di(p-tolyl)phenylamine (DTBT–TPA), respectively, tune the hole mobility of the polymer and enhance the performance of the PSCs with increased hole mobility (Fig. 2).31
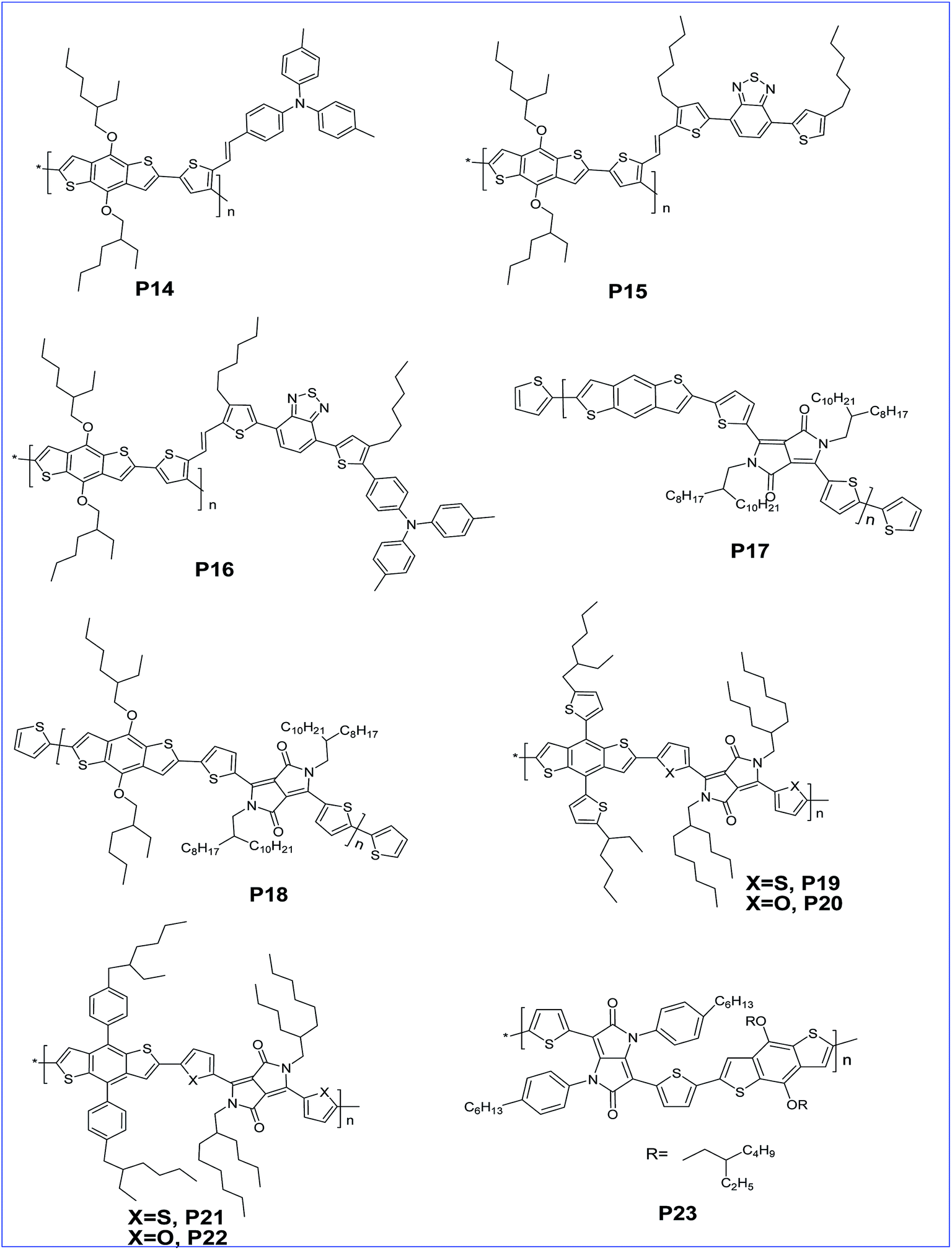 | ||
| Fig. 2 BDT based polymers with different side chains such as TPA, DTBT, DTBT–TPA and DPP (P14–P23). | ||
Diketopyrrolo[3,4-c]pyrrole (DPP) is an another acceptor unit copolymerised along with BDT for the preparation of various conjugated copolymers. Generally, it is a key structural unit found in an important class of red pigments. It was shown that DPP-containing polymers exhibit good light-emitting and photovoltaic properties.32–35 It is an electron deficient molecule with high conjugation and strong π–π interactions which makes it a viable building block for PSCs.36–38 The photovoltaic property of this acceptor was verified with many donor moieties and especially with BDT derivatives. Jae et al. synthesised a D–A copolymer of un-substituted BDT and DPP. The removal of the alkoxy side group from P17 not only lowers the HOMO level but also raises the Voc to 0.82 V with a PCE of 5.16%, whereas the alkoxy substituted P18 has a Voc of 0.61 V and a PCE of 2.24%.39 Yang and co-workers prepared copolymers P19, P20, P21 and P22 containing alkylthienyl- or alkylphenyl-substituted BDT units via Stille coupling. Single junction solar cells of the polymers showed very good PCEs in the range of 5–6%. Further, the performance is improved to 8% with tandem solar cells.40
Tetraaryl-diketopyrrolo[3,2-b]-pyrrole (isoDPP) is a regioisomer of DPP and was used as an acceptor with BDT to afford D–A polymer P23 by Yang and co-workers.41 The polymer exhibits a low band gap of 1.67 eV and a very broad absorption band ranging from 400 nm to 800 nm, which might be advantageous for solar cell applications since the absorption is extended to the region where the solar photon flux is most intense (that is in the 600–800 nm range).42,43 Further, a narrow optical band gap of nearly 1.64 eV for co-polymer P24 of thieno[3,2-b]thiophene diketopyrrolopyrrole and BDT was reported by Tandy et al. (Fig. 3). Here, the spacer in the polymer facilitates π–π stacking in the solid films. Due to this, the polymer has a hole and electron mobility of 10−3 cm2 V−1 s−1 and 10−5 cm2 V−1 s−1, respectively.44
 | ||
| Fig. 3 BDT based polymers with different side chains such as BDT, PDI, TDP, DTBT and DTffBT (P24–P37). | ||
The presence of a very good electron deficient moiety in the polymer potentially leads to an n-type polymer which actually acts as an acceptor of electrons compared to the donor polymer. This type of n-type polymers can potentially replace the fullerene acceptor. In fact, the combination of p-type and n-type macromolecules will have a better miscibility leading to improved morphology which will eventually increase device performance.45–49 Perylenediimide is one such building block utilised to synthesise a highly electron deficient semiconductor for synthesising n-type polymers.50
In this context, Li and colleagues synthesised n-type two-dimensional (2D)-conjugated polymer P25 based on bithienyl-benzodithiophene (bithienyl-BDT) and perylene diimide (PDI), via a Stille coupling reaction for application as an acceptor material in polymer solar cells (PSCs). The polymer P25 exhibits broad absorption in the visible region with an optical band gap of 1.64 eV, and a LUMO level of −3.89 eV which is similar to the energy level of PCBM, indicating that the polymer is suitable for application as an acceptor instead of PCBM in PSCs, and delivered a decent PCE of 4.71% for the device having a thienyl-BDT based polymer as the donor and P25 as the acceptor molecule.51 In another report, Kim et al. used a thienyl-BDT based polymer as the donor and a naphthalene diimide (NDI)-based polymer as the acceptor for polymer solar cell applications and observed an improved morphology, charge transfer and a PCE of 4.25% in the device.52
Hwang et al. synthesised a thieno[3,4-c]pyrrole-4,6-dione (TPD) based copolymer with bithiophene with a low HOMO and an impressive PCE of 9.21%.53 In addition, the TPD acceptor structure resembles that of DPP and the copolymer of TPD with BDT has also been explored.18 Recently, Zhu and co-workers prepared a copolymer (P26) containing TPD and BDT along with a thienothiophene (TT) linker. Here, the TT linker helps to reduce the band gap of polymer P26 and the polymer exhibited a very high Jsc of 18.2 mA cm−2 with a decent PCE of 7.5%.54
Benzothiadiazole (BT) is one of the strong electron withdrawing moieties widely used in PSCs and reflects their electron accepting strength as well as their capability to adopt the quinoid structure in the polymer, resulting in a low-band gap and more planar polymer. The incorporation of an electron withdrawing atom like fluorine has been proven to improve the performance of the polymer.55 Ho Jo et al. synthesised a fluorinated BT based conjugated polymer with lower HOMO and LUMO energy levels and an impressive PCE of 9.14%.56 In this context, two new D–A copolymers (P27 and P28) containing conjugated substituents like 4,7-dithien-5-yl-2,1,3-benzodiathiazole (DTBT) and 5,6-difluoro-4,7-bis(4-hexylthiophen-2-yl)benzo[c][1,2,5]thiadiazole (DTffBT) on thiazole were successfully synthesised by Tan and co-workers. Here, the presence of a conjugated side chain lowered the HOMO of both the polymers. Among the polymers, the fluorinated polymer P28 exhibited a better PCE of 3.75% compared to non-fluorinated analogue P27 with 2.63% PCE.57 Further, the incorporation of fluorine on BDT based copolymer P29 gave very low frontier orbitals which increased the Voc to 0.90 V and thus resulted in a PCE of 8.55%.58 Incorporating dithienylbenzothiadiazole–vinylene side chains into BDT based copolymers improves the film forming ability along with improved miscibility.59 In fact, bithienyl units as π-bridges are reported to enhance photovoltaic performances of conjugated polymers60 and particularly, 4,4′-bis(alkylsulfanyl)bithiophene was employed to promote better solubility.61
A copolymer of BDT and TT was reported to have a very good photovoltaic property. Yu et al. synthesised a copolymer of BDT and TT with different alkoxy side chains to afford P30 and its BHJ device delivered a PCE of 5.9%.62 Further, TT substituted BDT creates an extended π-system in the electron-donor moiety and in turn reduces the band gap of the polymers. Copolymer P31 of thieno[3,2-b]thiophenylbenzo[1,2-b:4,5-b′]dithiophene and 2-ethylhexyl 3-fluorothieno[3,4-b]thiophene-2-carboxylate (FTT) was synthesised having a low band gap of 1.55 eV and extended π-conjugation resulted in an efficiency of 7.44%.63
Shin et al. synthesised four copolymers P32, P33, P34 and P35 having both symmetrically and asymmetrically substituted alkoxy chains at positions 4 and 8 of BDT and concluded that the asymmetric polymer containing 4-octyl-8-octyloxy-BDT shows a better performance with 7.64% PCE.64 However, 1D conjugated polymer P30 was further modified, by replacing the alkoxy side chain with 5-alkylthiophene-2-yl side chains, resulting in a 2D conjugated polymer, P36. The introduction of 2D conjugation improved the charge mobility due to efficient π–π stacking of the polymer chains. Consequently, polymer P36 lead to a very low band gap, Voc and delivered a very high PCE of 10.12% for a single junction solar cell.65 The alkylthiophene-2-yl side chains on BDT were replaced by (E)-5-(2-(5-(alkylthio)thiophen-2-yl)vinyl)-thiophene-2-yl side chains to afford another 2D copolymer P37 by Hou et al. Copolymer P37 exhibited a wide absorption range and Voc of 0.81 V with a PCE of 8.13%.66 Chakravarthi and co-workers investigated the effect of changing the 2D conjugation. Wherein, 2D conjugation extension pathways were established in copolymer P38 by fusing the thiophenes to the phenyl core of 4,8-dithienylbenzo[1,2-b:4,5-b′]dithiophene. The polymer possesses less delocalized conjugation along the dithienylbenzene direction than in the BDT direction, and hence the PV property was reduced with a PCE of 7.1% for copolymer P38 (Fig. 4).67
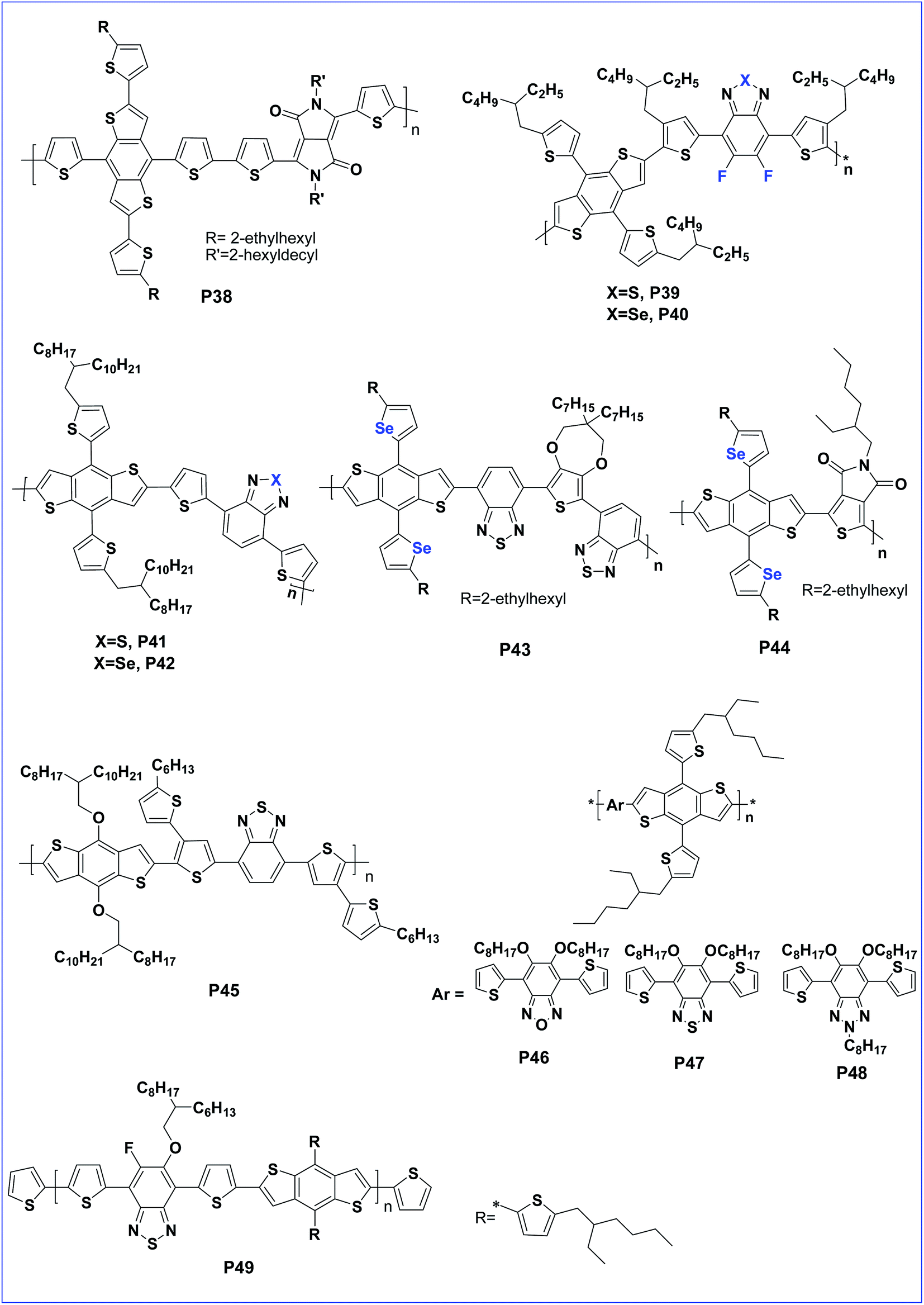 | ||
| Fig. 4 BDT based polymers with different side chains such as fused thiophenes, BDT, BT, BSe, DTBT, BBT, TPD and BDTSe (P38–P49). | ||
Two D–A copolymers (P39 and P40) based on bithienyl-benzodithiophene (bithienyl-BDT) as a donor (D) unit, and fluorinated benzothiadiazole (BT) or benzoselenadiazole (BSe) as an acceptor (A) unit, were designed and synthesized by Shen et.al. Copolymer P39 with an electron withdrawing fluorine atom performs better than copolymer P40 which also has poor morphology of the active layer.68 Further, the replacement of an S atom of diethyenyl benzothiazole by an Se atom, followed by copolymerisation with a BDT unit and with the addition of DIO as an additive during the device investigation, gives a more improved property. Furthermore, P41 and P42 showed an improved photovoltaic property of the copolymer with Se incorporated DTBT. The PCE of P41 was 5.01% whereas that of P42 was 5.18%.69 Yu et al. showed that replacing the sulphur with selenium increased the charge carrier mobility, reduced the band gap and achieved high a PCE of 6.87%.70 Further, an electron rich BDTSe moiety was prepared by incorporating 2-ethylhexylselenophene as a side chain substituent on positions 4 and 8 of BDT. Later, BDTSe was copolymerised with bisbenzothiadazole (BBT) and thienopyrroledione (TPD) to afford to D–A copolymers, P43 and P44, respectively. Copolymer P43 having BBT which is a good acceptor exhibited a PCE value of 4.51% whereas, P44 delivered 4.16% for the device.71
Benzothiadiazole (BT) is another acceptor building block which can lower the HOMO and LUMO of the copolymer. A copolymer of BDT with BT was synthesised having a hexylthiophene bridge, by Junzhen and co-workers, to afford P45. The polymer has a lower HOMO as anticipated and delivered a better PCE of 6.19%.72 A BT copolymer of BDT with alkylthienyl side groups was also reported to improve the performance of the polymer.73,74 Alkylthienyl substituted BDT was copolymerised with three different acceptors namely benzo-oxadiazole, benzothiadiazole and benzotriazole to afford copolymers P46, P47and P48, respectively. Among them, polymer P46, bearing benzo-oxadiazole delivered a better PCE of 5.9% due to its better acceptor property.75 The incorporation of a fluorine atom to benzothiadiazole and copolymerisation with alkylthienyl substituted BDT afforded P49. The polymer showed an improved morphology and formation of nano-fibres for a polymer:fullerene blend film, and hence an improved PCE of 6.88%.76
The performance can be tuned with the variation of oligothienyl side chains on the polymer to afford polymers P50, P51, P52 and P53 with extended π-conjugation side chains (Fig. 5). This π-extension improved the performance of P52 with a PCE of 6.48%.77 Incorporation of a π-bridge in the main polymer improved the optoelectronic properties of the polymer due to increasing the charge transfer ability.78 An alkylthienyl-substituted BDT copolymer with a 3-hexylthieno[3,2-b]thiophene π-bridge was reported to deliver a high PCE of 7.71% as anticipated.79 BDT substituted BDT (crossed BDT) possesses an enlarged planarity and is a very electron rich donor. Copolymerisation of this crossed BDT with benzothiadiazole was designed with or without a fluorine atom to obtain P54 and P55, respectively. The presence of very bulky BDT side groups is not efficient for molecular stacking and leads to a lower performance compared to alkylthienyl substituted polymers. However, compared to the non-fluorinated analogue, the fluorinated polymer, P55 was endowed with a lower HOMO resulting in a higher Voc.80
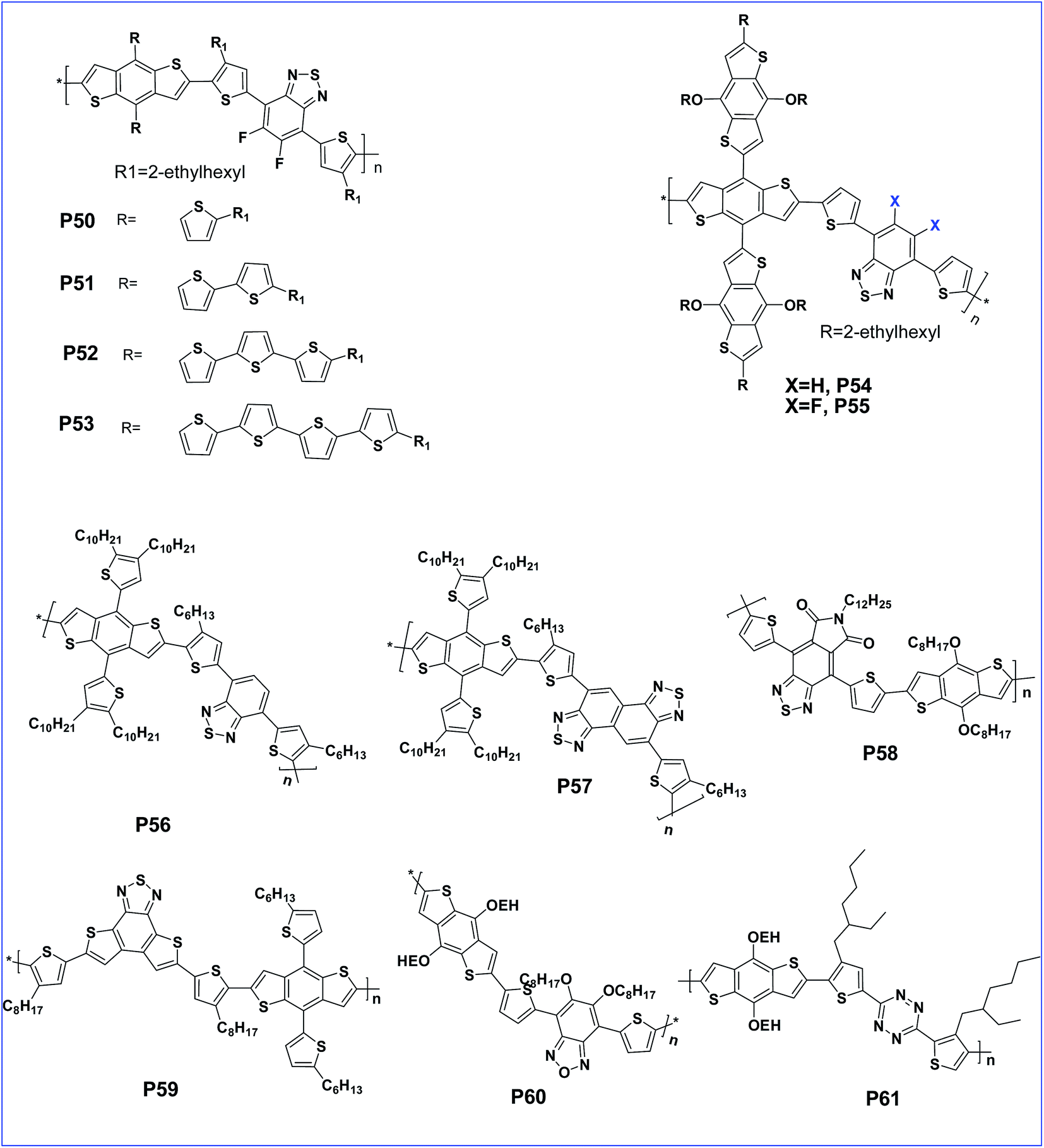 | ||
| Fig. 5 BDT based polymers with various alkylthienyl side chains (P50–P61). | ||
Cao et al. synthesised alkylthienyl-substituted BDT copolymers P56 and P57, based on 2,1,3-benzothiadiazole (BT) and naphtho[1,2-c:5,6-c]bis[1,2,5]thiadiazole (NT), respectively. The polymer bearing the NT unit, having a more quinoid structure, showed a reduced band gap and an improved efficiency of 6.00% compared to the BT analogue.81 Further, copolymer P58 carrying pyrrolo[3,4-f]-2,1,3-benzothiadiazole-5,7-dione (BTI) as the acceptor and BDT has a lower band gap and low lying HOMO energy levels.82 Another D–A copolymer of BDT with fused building block benzothiadiazole[1,2-b:4,3-b′] dithiophene (BTDT) (P59) creates a lower HOMO and hence exhibited a high open-circuit voltage (Voc) of 0.96 V and a moderate power conversion efficiency (PCE) of 2.16%.83
Benzooxadiazole (BO) has a much lower oxidation potential than BT due to the presence of an oxygen atom in place of sulfur.84 This leads to a very high Voc but it has a lower solubility84 which can be improved with the introduction of two octyloxy chains on the BO unit. In this context, a D–A polymer (P60) of BDT and 5,6-bis(octyloxy)benzo[c][1,2,5]-oxadiazole (BO) with alkoxy side chains showed a very low HOMO of −5.27 eV and exhibited a decent device performance with a PCE of 5.7%.85 Another D–A polymer (P61) based on bisthienyl-s-tetrazine (TTz) and BDT has a lower HOMO as well as increased thermal stability of the polymer thin film. Thus, the low lying HOMO improved the stability of the polymer.86
The introduction of an acceptor as the side chain facilitates hole mobility than main chain D–A copolymers.87 The property hence can be tuned with this design. In this context, a side chain acceptor polymer of BDT with a DPP acceptor side chain, P62 was designed which has an impressive hole mobility of 3.8 × 10−4 cm2 V−1 s−1 (Fig. 6).88,89
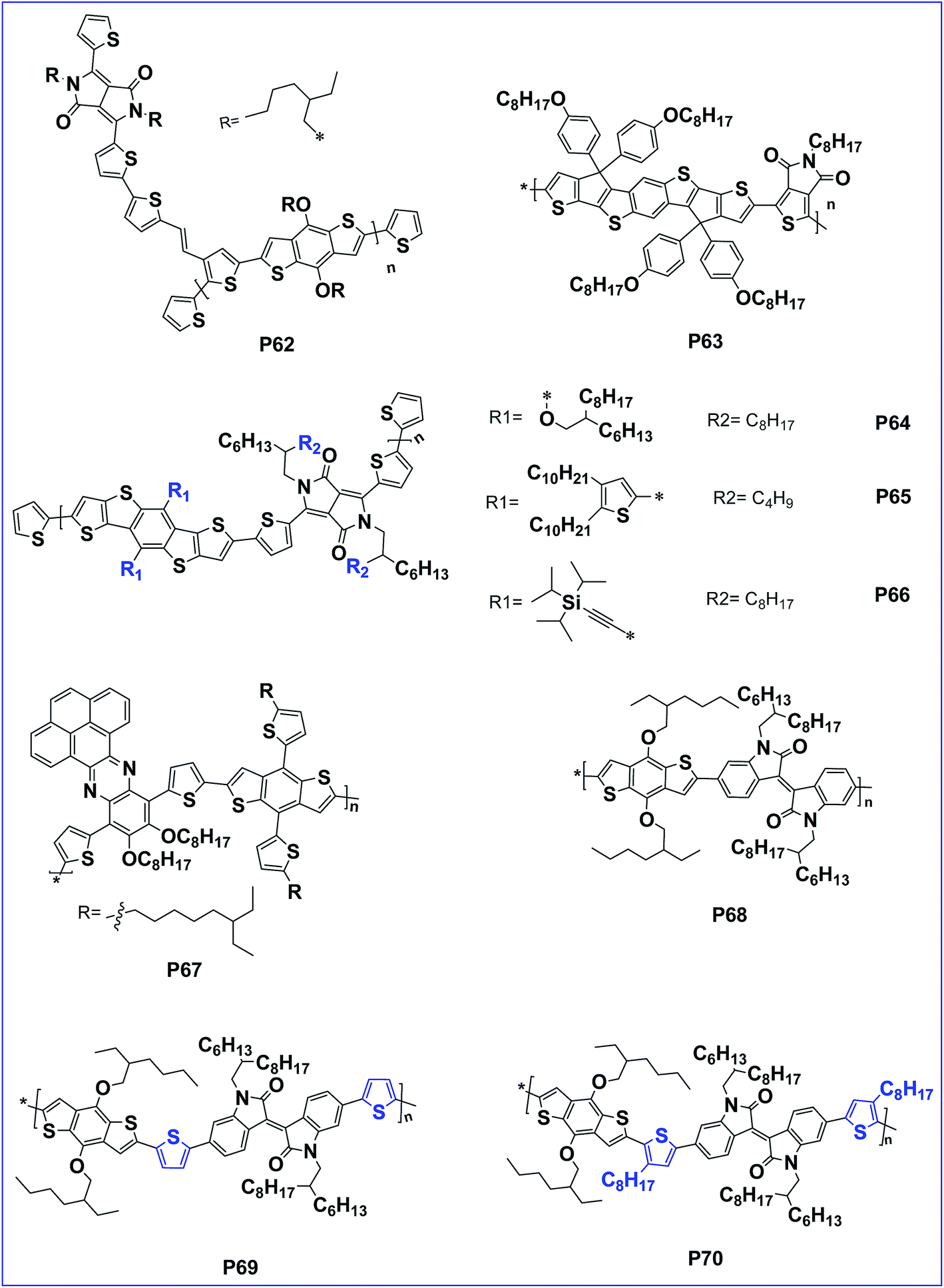 | ||
| Fig. 6 BDT based polymers with different side chains such as BDCPDT, DTBDT and PPz (P62–P70). | ||
Leclerc et al. first reported the synthesis of a copolymer based on BDT and thienopyrrole 4,6-dione. The solar cell with this polymer reached a power conversion efficiency of 5.5%.90 Further, to optimize light-harvesting and charge transporting abilities, two additional 3-octylthiophene units were introduced to the two sides of the BDT cores in the polymeric backbone. Unfortunately, the copolymer showed a reduced performance.91 Here, the poor performance of the polymer is attributed to inter-annular twisting of the polymer chain owing to the steric hindrance of the chain. To overcome this drawback, a distannyl-BDCPDT building block was copolymerized with 1,3-dibromo-thieno[3,4-c]pyrrole-4,6-dione (TPD) by Stille polymerization to afford alternating donor–acceptor copolymer P63 by Chen et.al. The presence of forced planarization in the polymer greatly suppresses the interannular twisting to extend the effective conjugated length and preserve the interactions between the donor and acceptor segments. Because of the planar configuration, P63 afforded an improved performance of 6.1%.92
Dithieno[2,3-d:2′,3′-d′]-benzo[1,2-b:4,5-b′]dithiophene (DTBDT) is an analogue of benzo[1,2-b:4,5-b′]dithiophene. It exhibits a similar HOMO level to BDT and has a larger coplanar structure facilitating charge-carrier mobility.93–95 In this context, the optoelectronic properties of conjugated polymers derived from DTBDTs and DPP (P64, P65 and P66) could effectively be tuned via flanking of DTBDTs by alkyloxy, 4,5-didecylthieno-2-yl and trialkyl silylethynyl substituent groups. Additionally, polymer P66 of tri-iso-propylsilylethynyl flanked DTBDTs exhibited a greater PCE of 6.39% when compared to the polymers of alkyloxy-flanked DTBDT and 4,5-didecylthieno-2-yl-flanked DTBDT.96
In the literature, some of the phenazine (Pz) derivatives are also utilized as an electron deficient (acceptor) units and showed very promising properties for application in PSCs. In general, they have a relatively large conjugated system with a planar structure that is usually favourable for strong inter molecular π–π stacking and self-assembling characteristics. These merits are beneficial for effective charge transportation and absorption shifts to longer wavelengths in the solar spectrum.97 In this perspective, D–A conjugated polymer P67, bearing pyrene-fused phenazine (PPz) and 7,8-bithienyl benzo[1,2-b:4,5-b0]dithiophene (BDTT), is found to have an extended planar structure with enhanced hole mobility. Because of the increased inter-chain π–π interactions, an optimized P67/PC71BM device showed a PCE of 4.86%.98
Isoindigo motif is well recognized as an electron-accepting building block for the preparation of electro-active materials for organo-electronics. Its high yielding and scalable synthesis enabled the rapid development of a large number of molecular and polymeric isoindigo-based materials with remarkable physical properties. In this context, three alternating polymers (P68, P69 and P70) with the electron-deficient isoindigo and BDT flanked with thiophenes or octylthiophenes as the donor units were reported by Ma et al. These polymers possess good thermal stability, solubility and broad absorption spectra. Among these three polymers, polymer P69, with BDT flanked by thiophenes as the donor unit and isoindigo as the acceptor unit, exhibits quite planar backbones and its blend with fullerene derivatives showed optimal morphology. As a result, the PSCs based on P69 with a device configuration of ITO/PEDOT:PSS/P69:PC61BM/LiF/Al showed a PCE of 4.22%. In fact, the performance of P69 was even better than P70. The lower performance of P70 is attributed to the inefficient π–π stacking caused by the presence of the octyloxy group (Table 1).99
| Polymer | λmax (nm) | Eoptg (eV) | HOMO (eV) | LUMO (eV) | Eecg (eV) | Voc (V) | Jsc (mA cm−2) | FF (%) | PCE (%) | Device configuration |
|---|---|---|---|---|---|---|---|---|---|---|
| P1 | 483, 523 | 2.21 | −5.31 | −2.94 | 2.37 | 0.83 | 4.18 | 45 | 1.56 | ITO/PEDOT:PSS(30 nm)/active layer/Ca(10 nm)/Al(100 nm) |
| P2 | 505, 544 | 2.03 | −5.41 | −3.26 | 2.15 | 0.91 | 4.4 | 43 | 1.73 | ITO/PEDOT:PSS(30 nm)/active layer/Ca(10 nm)/Al(100 nm) |
| P3 | 502, 539 | 2.07 | −5.36 | −3.11 | 2.25 | 0.99 | 7.66 | 53 | 4 | ITO/PEDOT:PSS(30 nm)/active layer/Ca(10 nm)/Al(100 nm) |
| P4 | 549 | — | −5.14 | −3.28 | 1.82 | 0.81 | 4.71 | 28.7 | 1.09 | ITO/PEDOT:PSS(60 nm)/active layer/Ca(10 nm)/Al(100 nm) |
| P5 | 541 | — | −5.19 | −3.3 | 1.83 | 0.88 | 5.67 | 30.5 | 1.52 | ITO/PEDOT:PSS(60 nm)/active layer/Ca(10 nm)/Al(100 nm) |
| P6 | 548 | — | −5.19 | −3.38 | 1.81 | 0.87 | 4.42 | 50.8 | 1.96 | ITO/PEDOT:PSS/active layer/Ca/Al |
| P7 | 541 | — | −5.23 | −3.33 | 1.8 | 0.71 | 4.21 | 34 | 1.01 | ITO/PEDOT:PSS/active layer/Ca/Al |
| P8 | 557 | — | −5.27 | −3.39 | 1.79 | 0.81 | 4.93 | 55.2 | 2.21 | ITO/PEDOT:PSS/active layer/Ca/Al |
| P9 | — | — | −5.44 | −3.46 | 1.98 | 0.93 | 11.95 | 55 | 6.12 | ITO/(PEDOT:PSS)/active layer/LiF/Al |
| P10 | 652, 721 | 1.59 | −5.15 | −3.44 | 1.71 | 0.63 | 8.78 | 53.3 | 2.9 | ITO/(PEDOT:PSS)/active layer/LiF/Al |
| P11 | 690, 766 | 1.47 | −5.24 | −3.74 | 1.5 | 0.72 | 12.64 | 56.5 | 5.1 | ITO/(PEDOT:PSS)/active layer/LiF/Al |
| P12 | 423 | 1.75 | −5.22 | −3.40 | 1.82 | 0.71 | 7.19 | 49 | 2.48 | ITO/PEDOT:PSS/active layer/Ca/Al |
| P13 | 504 | 1.92 | −5.44 | −3.44 | 2 | 0.81 | 12.65 | 41 | 4.18 | ITO/PEDOT:PSS/active layer/Ca/Al |
| P14 | 399 | 2.02 | −5.35 | −3.33 | — | 0.58 | 9.17 | 32 | 1.56 | ITO/(PEDOT:PSS)/active layer/LiF/Al |
| P15 | 504 | 1.92 | −5.55 | −3.44 | — | 0.83 | 14.88 | 42 | 5.35 | ITO/(PEDOT:PSS)/active layer/LiF/Al |
| P16 | 497 | 1.84 | −5.38 | −3.54 | — | 0.77 | 14.26 | 47 | 5.16 | ITO/(PEDOT:PSS)/active layer/LiF/Al |
| P17 | 750 | 1.31 | −5.46 | −3.74 | 1.72 | 0.82 | 10.49 | 60 | 5.16 | ITO/(PEDOT:PSS)/active layer/LiF/Al |
| P18 | 740 | 1.31 | −5.29 | −3.55 | 1.74 | 0.61 | 9.16 | 40.2 | 2.24 | ITO/(PEDOT:PSS)/active layer/LiF/Al |
| P19 | 710, 769 | 1.44 | −5.3 | −3.36 | — | 0.73 | 14 | 65 | 6.6 | ITO/PEDOT:PSS/active layer/Ca/Al |
| P20 | 687, 753 | 1.46 | −5.35 | −3.56 | — | 0.76 | 13.6 | 60 | 6.2 | ITO/PEDOT:PSS/active layer/Ca/Al |
| P21 | 681, 757 | 1.55 | −5.26 | −3.64 | — | 0.77 | 13.8 | 55 | 5.8 | ITO/PEDOT:PSS/active layer/Ca/Al |
| P22 | 676, 752 | 1.52 | −5.33 | −3.57 | — | 0.77 | 7.42 | 59 | 3.3 | ITO/PEDOT:PSS/active layer/Ca/Al |
| P23 | 371, 509 | 1.55 | −5.08 | −3.41 | 1.67 | — | — | — | — | — |
| P24 | 776 | 1.64 | — | — | — | 0.59 | 9.6 | 55 | 3.2 | ITO/PEDOT:PSS/active layer/Al |
| P25 | 385, 547, 656 | 1.6 | −5.7 | −3.89 | — | 0.8 | 11.51 | 51.1 | 4.71 | ITO/PEDOT:PSS/active layer)/Ca/Al |
| P26 | 725 | 1.56 | −4.78 | — | — | 0.633 | 18.15 | 65.2 | 7.5 | ITO/PEDOT:PSS/active layer/Ca/Al |
| P27 | 510 | 1.91 | −5.58 | −3.65 | 1.93 | 0.76 | 7.75 | 45 | 2.63 | ITO/PEDOT:PSS(30 nm)/active layer(10 nm)/Al(100 nm) |
| P28 | 513 | 1.92 | −5.59 | −3.57 | 2.02 | 0.76 | 9.05 | 54 | 3.75 | ITO/PEDOT:PSS(30 nm)/active layer(10 nm)/Al(100 nm) |
| P29 | 618 | 1.73 | −5.35 | −3.55 | 1.8 | 0.9 | 13.52 | 70.02 | 8.55 | ITO/PEDOT:PSS/active layer/Ca/Al |
| P30 | — | 1.63 | −5.12 | −3.31 | 1.81 | 0.74 | 13 | 61.4 | 5.9 | ITO/PEDOT:PSS/active layer/Ca/Al |
| P31 | 700 | — | −5.31 | −3.76 | 1.55 | 0.78 | 14.2 | 64 | 7.44 | ITO/PEDOT:PSS/polymer:PC71BM/Ca/Al |
| P32 | 670 | 1.6 | −5.11 | −3.51 | — | 0.66 | 16.29 | 67 | 7.18 | ITO/PEDOT:PSS/active layer/Ca/Al |
| P33 | 668 | 1.61 | −5.24 | −3.63 | — | 0.73 | 15.23 | 69 | 7.64 | ITO/PEDOT:PSS/active layer/Ca/Al |
| P34 | 657 | 1.6 | −5.28 | −3.68 | — | 0.78 | 12.48 | 66 | 6.48 | ITO/PEDOT:PSS/active layer/Ca/Al |
| P35 | 659 | 1.62 | −5.2 | −3.58 | — | 0.73 | 15.78 | 71 | 8.12 | ITO/PEDOT:PSS/active layer/Ca/Al |
| P36 | 325, 708 | 1.59 | −5.49 | −3.74 | 1.75 | 0.79 | 19.78 | 65 | 10.12 | ITO/PEDOT:PSS/active layer/Al |
| P37 | 4.02, 665, 734 | 1.53 | −5.29 | — | — | 0.81 | 16 | 62.71 | 8.13 | ITO/PEDOT:PSS/active layer/Mg/Al |
| P38 | 634 | 1.4 | −5.28 | −3.88 | 1.4 | 0.81 | 14.63 | 60.51 | 7.1 | ITO/ZnO/active layer/PEDOT:PSS/Ag |
| P39 | 358, 424, 576 | 1.74 | −5.34 | −3.23 | 2.11 | 0.9 | 9.3 | 46.9 | 3.93 | ITO/PEDOT:PSS/active layer/Ca/Al |
| P40 | 344, 436, 606 | 1.66 | −5.3 | −3.47 | 1.83 | 0.79 | 5.56 | 34.7 | 1.53 | ITO/PEDOT:PSS/active layer/Ca/Al |
| P41 | 425, 630 | 1.72 | −5.26 | −3.5 | — | 0.72 | 11.16 | 62 | 5.01 | ITO/PEDOT:PSS/active layer/Ca/Al |
| P42 | 446, 673 | 1.55 | −5.18 | −3.48 | — | 0.6 | 13.58 | 64 | 5.18 | ITO/PEDOT:PSS/active layer/Ca/Al |
| P43 | 650 | 1.64 | −5.34 | −3.7 | 0.65 | 11.72 | 58.69 | 4.51 | ITO/PEDOT:PSS/active layer/LiF/Al | |
| P44 | 606 | 1.85 | −5.73 | −3.88 | 0.68 | 9.94 | 52.76 | 3.58 | ITO/PEDOT:PSS/active layer/LiF/Al | |
| P45 | 422, 588 | 1.73 | −5.03 | −2.71 | 2.32 | 0.8 | 12.56 | 60.38 | 6.06 | ITO/PEDOT:PSS/active layer/Ca/Al |
| P46 | 59 | 1.69 | −5.25 | −3.28 | 1.87 | 0.84 | 11.45 | 61.3 | 5.9 | ITO/PEDOT:PSS/active layer/Ca/Al |
| P47 | 583 | 1.66 | −5.15 | −3.3 | 1.85 | 0.75 | 10.29 | 64 | 4.94 | ITO/PEDOT:PSS/active layer/Ca/Al |
| P48 | 540 | 1.78 | −4.92 | −3.01 | 1.91 | 0.54 | 9.47 | 60.6 | 3.1 | ITO/PEDOT:PSS/active layer/Ca/Al |
| P49 | 442, 641 | 1.79 | −5.29 | −3.5 | — | 0.76 | 14.67 | 62 | 6.88 | ITO/PEDOT:PSS/active layer/LiF/Al |
| P50 | 343, 407, 548 | 1.75 | −5.47 | −3.58 | 1.89 | 0.98 | 9.63 | 59.7 | 5.46 | ITO/MoO3/active layer/LiF/Al |
| P51 | 317, 391, 587 | 1.73 | −5.37 | −3.61 | 1.76 | 0.9 | 10.6 | 54.7 | 5.22 | ITO/MoO3/active layer/LiF/Al |
| P52 | 410, 608 | 1.7 | −5.31 | −3.62 | 1.69 | 0.86 | 12.3 | 61.4 | 6.48 | ITO/MoO3/active layer/LiF/Al |
| P53 | 425, 608 | 1.68 | −5.30 | −3.63 | 1.67 | 0.84 | 6.35 | 40.5 | 2.17 | ITO/MoO3/active layer/LiF/Al |
| P54 | 627 | 1.69 | −5.26 | −3.53 | 1.73 | 0.91 | 7.54 | 54.56 | 3.74 | ITO/PEDOT:PSS/active layer/Ca/Al |
| P55 | 612 | 1.77 | −5.34 | −3.6 | 1.74 | 0.94 | 2.35 | 51.05 | 1.13 | ITO/PEDOT:PSS/active layer/Ca/Al |
| P56 | — | 1.73 | −5.26 | −3.1 | 2.16 | 1 | 5.8 | 34.6 | 2.11 | ITO/PEDOT:PSS/active layer/Ca/Al |
| P57 | — | 1.58 | −5.19 | −3.26 | 1.93 | 0.8 | 11.71 | 61 | 6 | ITO/PEDOT:PSS/active layer/Ca/Al |
| P58 | 610, 444, 392 | — | −5.36 | −3.68 | 1.68 | 0.75 | 9.8 | 47 | 3.42 | ITO/PEDOT:PSS/active layer/TioX/Al |
| P59 | 356 | 2.1 | −5.54 | −3.44 | — | 0.96 | 5.49 | 41 | 2.16 | ITO/PEDOT:PSS/active layer/LiF/Al |
| P60 | 415, 575 | 1.74 | −5.27 | −3.16 | 2.11 | 0.86 | 10.4 | 64.4 | 5.7 | ITO/PEDOT:PSS/active layer/Ca(30 nm)/Al(100 nm) |
| P61 | 440, 516 | 1.85 | −5.42 | −3.58 | 1.84 | 0.92 | 6.21 | 58 | 3.32 | ITO/PEDOT:PSS/active layer/LiF/Al |
| P62 | 427, 588 | 1.73 | −5.34 | −3.52 | 1.82 | 0.84 | 10.04 | 56 | 4.89 | ITO/PEDOT:PSS(40 nm)/active layer/MoO3/Al |
| P63 | 614 | 1.88 | −5.36 | −3.2 | 2.16 | 0.85 | 11.95 | 60.4 | 6.1 | ITO/PEDOT:PSS/active layer/Ca/Al |
| P64 | 405, 750 | 1.48 | −5.13 | −3.65 | — | 0.62 | 6.41 | 60.9 | 2.42 | ITO/PEDOT:PSS/active layer/Ca/Al |
| P65 | — | 1.47 | −5.19 | −3.75 | — | 0.65 | 12.61 | 63.4 | 5.19 | ITO/PEDOT:PSS/active layer/Ca/Al |
| P66 | — | 1.45 | −5.25 | −3.8 | — | 0.75 | 13.9 | 61.3 | 6.39 | ITO/PEDOT:PSS/active layer/Ca/Al |
| P67 | 450, 604 | 1.59 | −5.34 | −3.15 | 2.19 | 0.7 | 11.1 | 62.5 | 4.86 | ITO/PEDOT:PSS/active layer/Ca/Al |
| P68 | 685 | 1.58 | −5.82 | −3.88 | 1.94 | 0.84 | 3.9 | 46 | 1.53 | ITO/PEDOT:PSS/active layer/LiF/Al |
| P69 | 660 | 1.61 | −5.78 | −3.91 | 1.87 | 0.79 | 7.87 | 68 | 4.22 | ITO/PEDOT:PSS/active layer/LiF/Al |
| P70 | 635 | 1.59 | −5.74 | −3.91 | 1.83 | 0.73 | 3.15 | 60 | 1.39 | ITO/PEDOT:PSS/active layer/LiF/Al |
| P71 | 475 | 2.01 | −5.16 | −2.82 | 2.34 | 0.88 | 5.2 | 61.3 | 2.81 | ITO/PEDOT:PSS/active layer/Ca/Al |
| P72 | 485 | 1.91 | −5.19 | −2.9 | 2.29 | 0.9 | 5.5 | 68.7 | 3.4 | ITO/PEDOT:PSS/active layer/Ca/Al |
| P73 | 532 | 1.7 | −5.09 | −3.1 | 1.99 | 0.8 | 4.58 | 30.7 | 1.13 | ITO/PEDOT:PSS/active layer/Ca/Al |
| P74 | 373, 577 | 1.75 | −5.09 | −3.34 | — | 0.81 | 6.36 | 41 | 2.12 | ITO/PEDOT:PSS/active layer/Ca/Ag |
| P75 | 657 | 1.27 | −4.96 | −3.69 | — | 0.67 | 2.79 | 43 | 0.8 | ITO/PEDOT4083/active layer/Al |
| P76 | 532 | 2.05 | −5.65 | −3.6 | — | 0.81 | 10.9 | 57 | 5.06 | ITO/PEDOT:PSS/active layer/LiF/Al |
Zhang et al. prepared three D–A copolymers (P71, P72 and P73) containing a dioctyloxybenzo[1,2-b:4,3-b0]dithiophene (BdT) donor unit and different acceptor units of bithiazole (BTz), thiazolo[5,4-d]-thiazole (TTz) and dithienylylbenzothiadiazole (DTBT) with alkyl groups present at different positions on the main chain (Fig. 7). Actually, benzo[1,2-b:4,3-b′]dithiophene (BdT) is an isomer of BDT and possesses similar properties to BDT. Among these polymers, the PSCs based on the blend of P72 (with a HOMO level of −5.19 eV) and PC70BM (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 w/w) exhibited a greater PCE of 3.40%.100
:3 w/w) exhibited a greater PCE of 3.40%.100
 | ||
| Fig. 7 BDT based polymers with different side chains such as BdT, BTz, TTz and BTT (P71–P77). | ||
Quantum-chemical calculations suggest that the benzo[1,2-b:3,4-b′:5,6-d′′]trithiophene (BTT) unit possesses a similar donor strength ability in comparison with the well-known BDT unit. Further, an electron-rich fused aromatic unit (BTT) has a more planar configuration favouring π–π stacking compared to BDT. Further, BTT was copolymerised with electron deficient benzothidiazole (BT) as the acceptor to afford D–A polymer P74 having a band gap of 1.75 eV and a PCE of 2.2%, due to the poor donor capability of BTT.101 However, the band gap can be further reduced by copolymerizing benzotrithiophene (BTT) with diketopyrrolopyrrole (DPP) to afford P75 having a lower band gap of 0.80 eV with absorption spectra extending to the near-infrared region. Unfortunately, a lower PCE of 1.39% was achieved for this polymer due to the poor donor capability of BTT.102 To improve the performance of the BTT based polymer, the BTT unit was copolymerised with bithiazole (acceptor) to get a polymer (P76). Consequently, polymer P76 possesses a very low lying HOMO and a PSC device of P76:PCBM delivered a PCE of 2.49%. In addition, the performance of the PSC device was doubled (PCE to 5.06%) with the addition of a DIO additive which generally creates a nano scale morphology in the blend film.103
Recently, the electronic-rich subunit and planar 1,3-bis(5-bromothiophen-2-yl)-5,7-bis(2-ethylhexyl)-4H,8H-benzo[1,2-c:4,5-c′]dithiophene-4,8-dione as the electronic-deficient subunit were coupled with 2-ethylhexylphenyl substituted BDT to get copolymer P77. The PSC device of P77 with PCBM achieved a PCE of 3.62%. The active layer had a large aggregation with a domain size of 100 nm which was reduced to 20–30 nm with the addition of 1% DIO as a solvent additive, which also improved the efficiency to 8.0%, which is favorable for excitons to dissociate at the donor–acceptor interface and for improved efficiency.104
Conclusion and outlook
Over the past few years, various photovoltaic materials for PSC applications have been designed and developed to achieve a bench mark efficiency of 10%. To attain this, polymers based on BDT/structurally modified BDT (as a donor only type polymer) or BDT in conjunction with other acceptor moieties like NTDO, DPP, PDI, TPD, BT, BSe, BBT, BO, TTz, BTI, Pz, and isoindigo (as a D–A type polymer) are found to be an excellent strategy. Homopolymers of alkylthienyl substituted BDT have demonstrated very good performance due to 2D conjugation which extends the π-conjugation. However, the hole mobility in the polymer can be improved further with the introduction of a strong electron withdrawing building block, thereby improving the performance of PSCs. Stronger acceptor building blocks also assist in the reduction of the polymer. The solubility of the polymers are found to be attributed to the presence of side chains, however, bulky side chains were also found to hinder the performance since they hamper the molecular stacking. However, linkers, like thienothiophene, were found to improve the π–π interactions and hence reduce the band gap leading to a broader range of absorption in the visible region of the solar spectrum.Perylenediimide and naphthalene diimide building blocks along with thienyl substituted BDT polymers were found to be very good electron accepting materials and hence can be utilised as n-type polymer materials for the fabrication of BHJ devices with thienyl substituted BDT as donor, p-type materials. This kind of polymer acceptor can potentially replace the fullerene acceptor and hence, can improve the morphology of the active layer and is expected to deliver a stable BHJ device. Polymers based on alkylthienyl substituted BDT were found to have very interesting photoactive properties and hence are explored in recent times. As a whole, the planar conjugated nature, efficient π–π stacking ability, and the relatively high hole mobility of BDT units in the polymer tremendously contribute to the impressive photovoltaic properties.
Acknowledgements
This research was supported by the Solar Energy Research Initiative (Project code: DST/TMC/SERI/FR/201), Department of Science and Technology (DST), Govt. of India, New Delhi.References
- G. Yu, J. Gao, J. C. Hummelen, F. Wudl and A. J. Heeger, Science, 1995, 270, 1789–1791 CAS.
- J. You, L. Dou, K. Yoshimura, T. Kato, K. Ohya, T. Moriarty, K. Emery, C.-C. Chen, J. Gao, G. Li and Y. Yang, Nat. Commun., 2013, 4, 1446 CrossRef PubMed.
- Y. Li and Y. Zou, Adv. Mater., 2008, 20, 2952–2958 CrossRef CAS.
- F. G. Brunetti, R. Kumar and F. Wudl, J. Mater. Chem., 2010, 20, 2934–2948 RSC.
- M. Helgesen, R. Sondergaard and F. C. Krebs, J. Mater. Chem., 2010, 20, 36–60 RSC.
- P. Heremans, D. Cheyns and B. P. Rand, Acc. Chem. Res., 2009, 42, 1740–1747 CrossRef CAS PubMed.
- G. Li, V. Shrotriya, J. Huang, Y. Yao, T. Moriarty, K. Emery and Y. Yang, Nat. Mater., 2005, 4, 864–868 CrossRef CAS.
- R. D. McCullough, Adv. Mater., 1998, 10, 93–116 CrossRef CAS.
- M. Reyes-Reyes, K. Kim and D. L. Carroll, Appl. Phys. Lett., 2005, 87, 083506 CrossRef.
- J. Roncali, Chem. Rev., 1992, 92, 711–738 CrossRef CAS.
- S. Beaupré, P.-L. T. Boudreault and M. Leclerc, Adv. Mater., 2010, 22, E6–E27 CrossRef PubMed.
- S. H. Park, A. Roy, S. Beaupre, S. Cho, N. Coates, J. S. Moon, D. Moses, M. Leclerc, K. Lee and A. J. Heeger, Nat. Photonics, 2009, 3, 297–302 CrossRef CAS.
- P.-L. T. Boudreault, A. Najari and M. Leclerc, Chem. Mater., 2011, 23, 456–469 CrossRef CAS.
- E. Zhou, Q. Wei, S. Yamakawa, Y. Zhang, K. Tajima, C. Yang and K. Hashimoto, Macromolecules, 2010, 43, 821–826 CrossRef CAS.
- M. C. Scharber, D. Mühlbacher, M. Koppe, P. Denk, C. Waldauf, A. J. Heeger and C. J. Brabec, Adv. Mater., 2006, 18, 789–794 CrossRef CAS.
- C. Duan, K.-S. Chen, F. Huang, H.-L. Yip, S. Liu, J. Zhang, A. K. Y. Jen and Y. Cao, Chem. Mater., 2010, 22, 6444–6452 CrossRef CAS.
- Y. Huang, L. Huo, S. Zhang, X. Guo, C. C. Han, Y. Li and J. Hou, Chem. Commun., 2011, 47, 8904–8906 RSC.
- L. Huo and J. Hou, Polym. Chem., 2011, 2, 2453–2461 RSC.
- H. Zhou, L. Yang, S. Stoneking and W. You, ACS Appl. Mater. Interfaces, 2010, 2, 1377–1383 CAS.
- M.-C. Yuan, M.-Y. Chiu, C.-M. Chiang and K.-H. Wei, Macromolecules, 2010, 43, 6270–6277 CrossRef CAS.
- J. Hou, H.-Y. Chen, S. Zhang, R. I. Chen, Y. Yang, Y. Wu and G. Li, J. Am. Chem. Soc., 2009, 131, 15586–15587 CrossRef CAS PubMed.
- Y. Liang, Z. Xu, J. Xia, S.-T. Tsai, Y. Wu, G. Li, C. Ray and L. Yu, Adv. Mater., 2010, 22, E135–E138 CrossRef CAS PubMed.
- L. Huo, J. Hou, H.-Y. Chen, S. Zhang, Y. Jiang, T. L. Chen and Y. Yang, Macromolecules, 2009, 42, 6564–6571 CrossRef CAS.
- D. Lee, E. Hubijar, G. J. D. Kalaw and J. P. Ferraris, Chem. Mater., 2012, 24, 2534–2540 CrossRef CAS.
- C. Cui, H. Fan, X. Guo, M. Zhang, Y. He, X. Zhan and Y. Li, Polym. Chem., 2012, 3, 99–104 RSC.
- X. Chen, B. Liu, Y. Zou, W. Tang, Y. Li and D. Xiao, RSC Adv., 2012, 2, 7439 RSC.
- T. E. Kang, T. Kim, C. Wang, S. Yoo and B. J. Kim, Chem. Mater., 2015, 27, 2653–2658 CrossRef CAS.
- J. Hou, Z. a. Tan, Y. Yan, Y. He, C. Yang and Y. Li, J. Am. Chem. Soc., 2006, 128, 4911–4916 CrossRef CAS PubMed.
- Y. Wang, F. Yang, Y. Liu, R. Peng, S. Chen and Z. Ge, Macromolecules, 2013, 46, 1368–1375 CrossRef CAS.
- Z. Gu, P. Tang, B. Zhao, H. Luo, X. Guo, H. Chen, G. Yu, X. Liu, P. Shen and S. Tan, Macromolecules, 2012, 45, 2359–2366 CrossRef CAS.
- H. Li, H. Luo, Z. Cao, Z. Gu, P. Shen, B. Zhao, H. Chen, G. Yu and S. Tan, J. Mater. Chem., 2012, 22, 22913 RSC.
- E. Q. Guo, P. H. Ren, Y. L. Zhang, H. C. Zhang and W. J. Yang, Chem. Commun., 2009, 5859–5861, 10.1039/B911808J.
- B. Zhang, H. Zhang, X. Li, W. Li, P. Sun and W. Yang, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 3048–3057 CrossRef CAS.
- C. Yang, M. Zheng, Y. Li, B. Zhang, J. Li, L. Bu, W. Liu, M. Sun, H. Zhang, Y. Tao, S. Xue and W. Yang, J. Mater. Chem. A, 2013, 1, 5172–5178 CAS.
- Y. Zhu, A. R. Rabindranath, T. Beyerlein and B. Tieke, Macromolecules, 2007, 40, 6981–6989 CrossRef CAS.
- M. M. Wienk, M. Turbiez, J. Gilot and R. A. J. Janssen, Adv. Mater., 2008, 20, 2556–2560 CrossRef CAS.
- J. C. Bijleveld, A. P. Zoombelt, S. G. J. Mathijssen, M. M. Wienk, M. Turbiez, D. M. de Leeuw and R. A. J. Janssen, J. Am. Chem. Soc., 2009, 131, 16616–16617 CrossRef CAS PubMed.
- Y. Li, P. Sonar, L. Murphy and W. Hong, Energy Environ. Sci., 2013, 6, 1684–1710 CAS.
- J. W. Jung, J. W. Jo, F. Liu, T. P. Russell and W. H. Jo, Chem. Commun., 2012, 48, 6933–6935 RSC.
- L. Dou, J. Gao, E. Richard, J. You, C. C. Chen, K. C. Cha, Y. He, G. Li and Y. Yang, J. Am. Chem. Soc., 2012, 134, 10071–10079 CrossRef CAS PubMed.
- H. Zhang, I. Welterlich, J.-M. Neudörfl, B. Tieke, C. Yang, X. Chen and W. Yang, Polym. Chem., 2013, 4, 4682 RSC.
- H. Usta, G. Lu, A. Facchetti and T. J. Marks, J. Am. Chem. Soc., 2006, 128, 9034–9035 CrossRef CAS PubMed.
- G. Lu, H. Usta, C. Risko, L. Wang, A. Facchetti, M. A. Ratner and T. J. Marks, J. Am. Chem. Soc., 2008, 130, 7670–7685 CrossRef CAS PubMed.
- K. Tandy, G. K. Dutta, Y. Zhang, N. Venkatramaiah, M. Aljada, P. L. Burn, P. Meredith, E. B. Namdas and S. Patil, Org. Electron., 2012, 13, 1981–1988 CrossRef CAS.
- C. R. McNeill, Energy Environ. Sci., 2012, 5, 5653–5667 CAS.
- T. Earmme, Y.-J. Hwang, N. M. Murari, S. Subramaniyan and S. A. Jenekhe, J. Am. Chem. Soc., 2013, 135, 14960–14963 CrossRef CAS PubMed.
- H. Li, T. Earmme, G. Ren, A. Saeki, S. Yoshikawa, N. M. Murari, S. Subramaniyan, M. J. Crane, S. Seki and S. A. Jenekhe, J. Am. Chem. Soc., 2014, 136, 14589–14597 CrossRef CAS PubMed.
- D. Mori, H. Benten, I. Okada, H. Ohkita and S. Ito, Adv. Energy Mater., 2014, 4(3), 1301006 Search PubMed.
- Y.-J. Hwang, T. Earmme, B. A. E. Courtright, F. N. Eberle and S. A. Jenekhe, J. Am. Chem. Soc., 2015, 137, 4424–4434 CrossRef CAS PubMed.
- X. Zhan, Z. a. Tan, B. Domercq, Z. An, X. Zhang, S. Barlow, Y. Li, D. Zhu, B. Kippelen and S. R. Marder, J. Am. Chem. Soc., 2007, 129, 7246–7247 CrossRef CAS PubMed.
- Y. Zhang, Q. Wan, X. Guo, W. Li, B. Guo, M. Zhang and Y. Li, J. Mater. Chem. A, 2015, 3, 18442–18449 CAS.
- J. Choi, K.-H. Kim, H. Yu, C. Lee, H. Kang, I. Song, Y. Kim, J. H. Oh and B. J. Kim, Chem. Mater., 2015, 27(15), 5230–5237 CrossRef CAS.
- J.-H. Kim, J. B. Park, I. H. Jung, A. C. Grimsdale, S. C. Yoon, H. Yang and D.-H. Hwang, Energy Environ. Sci., 2015, 8, 2352–2356 CAS.
- C. Zhang, H. Li, J. Wang, Y. Zhang, Y. Qiao, D. Huang, C.-a. Di, X. Zhan, X. Zhu and D. Zhu, J. Mater. Chem. A, 2015, 3, 11194–11198 CAS.
- S. Park, J. Cho, M. J. Ko, D. S. Chung and H. J. Son, Macromolecules, 2015, 48, 3883–3889 CrossRef CAS.
- J. W. Jo, J. W. Jung, E. H. Jung, H. Ahn, T. J. Shin and W. H. Jo, Energy Environ. Sci., 2015, 8, 2427–2434 CAS.
- X. Liu, Y. Huang, Z. Cao, C. Weng, H. Chen and S. Tan, Polym. Chem., 2013, 4, 4737 RSC.
- D. Liu, W. Zhao, S. Zhang, L. Ye, Z. Zheng, Y. Cui, Y. Chen and J. Hou, Macromolecules, 2015, 48(15), 5172–5178 CrossRef CAS.
- Z. Gu, P. Shen, S. W. Tsang, Y. Tao, B. Zhao, P. Tang, Y. Nie, Y. Fang and S. Tan, Chem. Commun., 2011, 47, 9381–9383 RSC.
- M. Zhang, X. Guo and Y. Li, Macromolecules, 2011, 44, 8798–8804 CrossRef CAS.
- P. Morvillo, F. Parenti, R. Diana, C. Fontanesi, A. Mucci, F. Tassinari and L. Schenetti, Sol. Energy Mater. Sol. Cells, 2012, 104, 45–52 CrossRef CAS.
- Y. Liang, D. Feng, Y. Wu, S.-T. Tsai, G. Li, C. Ray and L. Yu, J. Am. Chem. Soc., 2009, 131, 7792–7799 CrossRef CAS PubMed.
- J.-H. Kim, C. E. Song, B. Kim, I.-N. Kang, W. S. Shin and D.-H. Hwang, Chem. Mater., 2014, 26, 1234–1242 CrossRef CAS.
- Q. V. Hoang, C. E. Song, S.-J. Moon, S. K. Lee, J.-C. Lee, B. J. Kim and W. S. Shin, Macromolecules, 2015, 48, 3918–3927 CrossRef CAS.
- C. Liu, C. Yi, K. Wang, Y. Yang, R. S. Bhatta, M. Tsige, S. Xiao and X. Gong, ACS Appl. Mater. Interfaces, 2015, 7, 4928–4935 CAS.
- H. Yao, H. Zhang, L. Ye, W. Zhao, S. Zhang and J. Hou, Macromolecules, 2015, 48, 3493–3499 CrossRef CAS.
- N. Chakravarthi, K. Gunasekar, C. S. Kim, D.-H. Kim, M. Song, Y. G. Park, J. Y. Lee, Y. Shin, I.-N. Kang and S.-H. Jin, Macromolecules, 2015, 48, 2454–2465 CrossRef CAS.
- P. Shen, H. Bin, Y. Zhang and Y. Li, Polym. Chem., 2014, 5, 567–577 RSC.
- E. Zhou, J. Cong, K. Hashimoto and K. Tajima, Macromolecules, 2013, 46, 763–768 CrossRef CAS.
- H. A. Saadeh, L. Lu, F. He, J. E. Bullock, W. Wang, B. Carsten and L. Yu, ACS Macro Lett., 2012, 1, 361–365 CrossRef CAS.
- N. Chakravarthi, K. Kranthiraja, M. Song, K. Gunasekar, P. Jeong, S.-J. Moon, W. Suk Shin, I.-N. Kang, J. W. Lee and S.-H. Jin, Sol. Energy Mater. Sol. Cells, 2014, 122, 136–145 CrossRef CAS.
- J. Ren, X. Bao, L. Han, J. Wang, M. Qiu, Q. Zhu, T. Hu, R. Sheng, M. Sun and R. Yang, Polym. Chem., 2015, 6, 4415–4423 RSC.
- H.-Y. Chen, J. Hou, S. Zhang, Y. Liang, G. Yang, Y. Yang, L. Yu, Y. Wu and G. Li, Nat. Photonics, 2009, 3, 649–653 CrossRef CAS.
- Q. Peng, X. Liu, D. Su, G. Fu, J. Xu and L. Dai, Adv. Mater., 2011, 23, 4554–4558 CrossRef CAS PubMed.
- B. Liu, X. Chen, Y. He, Y. Li, X. Xu, L. Xiao, L. Li and Y. Zou, J. Mater. Chem. A, 2013, 1, 570–577 CAS.
- G. Li, B. Zhao, C. Kang, Z. Lu, C. Li, H. Dong, W. Hu, H. Wu and Z. Bo, ACS Appl. Mater. Interfaces, 2015, 7, 10710–10717 CAS.
- J. Lee, J.-H. Kim, B. Moon, H. G. Kim, M. Kim, J. Shin, H. Hwang and K. Cho, Macromolecules, 2015, 48, 1723–1735 CrossRef CAS.
- C. Gao, L. Wang, X. Li and H. Wang, Polym. Chem., 2014, 5, 5200–5210 RSC.
- S. Liu, X. Bao, W. Li, K. Wu, G. Xie, R. Yang and C. Yang, Macromolecules, 2015, 48, 2948–2957 CrossRef CAS.
- D. Liu, C. Gu, M. Xiao, M. Qiu, M. Sun and R. Yang, Polym. Chem., 2015, 6, 3398–3406 RSC.
- M. Wang, X. Hu, P. Liu, W. Li, X. Gong, F. Huang and Y. Cao, J. Am. Chem. Soc., 2011, 133, 9638–9641 CrossRef CAS PubMed.
- H. Li, S. Sun, S. Mhaisalkar, M. T. Zin, Y. M. Lam and A. C. Grimsdale, J. Mater. Chem. A, 2014, 2, 17925–17933 CAS.
- S. Hu, X. Bao, Z. Liu, T. Wang, Z. Du, S. Wen, N. Wang, L. Han and R. Yang, Org. Electron., 2014, 15, 3601–3608 CrossRef CAS.
- N. Blouin, A. Michaud, D. Gendron, S. Wakim, E. Blair, R. Neagu-Plesu, M. Belletête, G. Durocher, Y. Tao and M. Leclerc, J. Am. Chem. Soc., 2008, 130, 732–742 CrossRef CAS PubMed.
- J. M. Jiang, P. A. Yang, H. C. Chen and K. H. Wei, Chem. Commun., 2011, 47, 8877–8879 RSC.
- S. Wen, Q. Dong, W. Cheng, P. Li, B. Xu and W. Tian, Sol. Energy Mater. Sol. Cells, 2012, 100, 239–245 CrossRef CAS.
- F. Huang, K.-S. Chen, H.-L. Yip, S. K. Hau, O. Acton, Y. Zhang, J. Luo and A. K. Y. Jen, J. Am. Chem. Soc., 2009, 131, 13886–13887 CrossRef CAS PubMed.
- C. Duan, X. Hu, K.-S. Chen, H.-L. Yip, W. Li, F. Huang, A. K. Y. Jen and Y. Cao, Sol. Energy Mater. Sol. Cells, 2012, 97, 50–58 CrossRef CAS.
- H. Tan, X. Deng, J. Yu, B. Zhao, Y. Wang, Y. Liu, W. Zhu, H. Wu and Y. Cao, Macromolecules, 2013, 46, 113–118 CrossRef CAS.
- Y. Zou, A. Najari, P. Berrouard, S. Beaupré, B. Réda Aïch, Y. Tao and M. Leclerc, J. Am. Chem. Soc., 2010, 132, 5330–5331 CrossRef CAS PubMed.
- A. Najari, S. Beaupré, P. Berrouard, Y. Zou, J.-R. Pouliot, C. Lepage-Pérusse and M. Leclerc, Adv. Funct. Mater., 2011, 21, 718–728 CrossRef CAS.
- Y.-L. Chen, C.-Y. Chang, Y.-J. Cheng and C.-S. Hsu, Chem. Mater., 2012, 24, 3964–3971 CrossRef CAS.
- Y. Wu, Z. Li, X. Guo, H. Fan, L. Huo and J. Hou, J. Mater. Chem., 2012, 22, 21362 RSC.
- H. J. Son, L. Lu, W. Chen, T. Xu, T. Zheng, B. Carsten, J. Strzalka, S. B. Darling, L. X. Chen and L. Yu, Adv. Mater., 2013, 25, 838–843 CrossRef CAS PubMed.
- Y. Wu, Z. Li, W. Ma, Y. Huang, L. Huo, X. Guo, M. Zhang, H. Ade and J. Hou, Adv. Mater., 2013, 25, 3449–3455 CrossRef CAS PubMed.
- S. Sun, P. Zhang, J. Li, Y. Li, J. Wang, S. Zhang, Y. Xia, X. Meng, D. Fan and J. Chu, J. Mater. Chem. A, 2014, 2, 15316 CAS.
- K.-S. Chen, H.-L. Yip, C. W. Schlenker, D. S. Ginger and A. K. Y. Jen, Org. Electron., 2012, 13, 2870–2878 CrossRef CAS.
- Q. Fan, Y. Liu, M. Xiao, H. Tan, Y. Wang, W. Su, D. Yu, R. Yang and W. Zhu, Org. Electron., 2014, 15, 3375–3383 CrossRef CAS.
- Z. Ma, E. Wang, M. E. Jarvid, P. Henriksson, O. Inganäs, F. Zhang and M. R. Andersson, J. Mater. Chem., 2012, 22, 2306–2314 RSC.
- B. Liu, X. Chen, Y. Zou, Y. He, L. Xiao, X. Xu, L. Li and Y. Li, Polym. Chem., 2013, 4, 470–476 RSC.
- C. B. Nielsen, B. C. Schroeder, A. Hadipour, B. P. Rand, S. E. Watkins and I. McCulloch, J. Mater. Chem., 2011, 21, 17642 RSC.
- L. Bian, J. Miao, J. Hai, E. Zhu, J. Yu, G. Ge, H. Wu and W. Tang, RSC Adv., 2014, 4, 53939–53945 RSC.
- X. Zhao, D. Yang, H. Lv, L. Yin and X. Yang, Polym. Chem., 2013, 4, 57–60 RSC.
- L. Huo, T. Liu, B. Fan, Z. Zhao, X. Sun, D. Wei, M. Yu, Y. Liu and Y. Sun, Adv. Mater., 2015, 27, 6969–6975 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |