
Optimized polarization build-up times in dissolution DNP-NMR using a benzyl amino derivative of BDPA†
Abstract
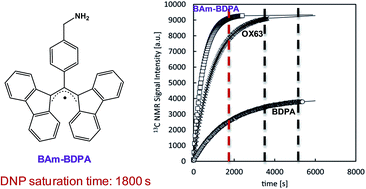
The synthesis of two novel BDPA-like radicals, a benzyl amino (BAm-BDPA, 7) and a cyano (CN-BDPA, 5) derivative, is reported and their behaviour as polarizing agents for fast dissolution Dynamic Nuclear Polarization (DNP) is evaluated. The radical 7 is a promising candidate for DNP studies since it is soluble in neat [1-13C]pyruvic acid (PA), and therefore the use of an additional glassing agent for sample homogeneity is avoided. In addition, a 60 mM sample of 7 offers optimum 13C NMR signal enhancements using fairly short polarization times (about 1800 s). It is shown that DNP-NMR measurements using 7 can be performed much more efficiently in terms of the signal enhancement per polarization build-up time unit than when using the reference OX63 or BDPA radicals. These enhanced features are translated to a substantial reduction of polarization times that represents an optimum temporary use of the DNP polarizer and allow economized liquid helium consumption.
 Please wait while we load your content...
Please wait while we load your content...

