
Computational-aided design of melatonin analogues with outstanding multifunctional antioxidant capacity†
 *a
*a
Abstract
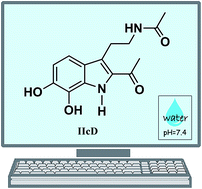
A set of 19 melatonin analogues, intended to be better antioxidants than the parent molecule, have been computationally designed. Eight of them were planned to have good primary antioxidant capacity (AOC), i.e., being good free radical scavengers. Seven of them were designed for their secondary AOC, being able to inhibit ˙OH production by acting as metal ion chelators. Based on their predicted behavior for the intended functions, four multifunctional melatonin analogues were proposed. They were found to be among the best peroxyl radical scavengers identified so far, in aqueous solution, at physiological pH. They were also found to be capable of turning off Cu(II) reduction by O2˙− and Asc−, thus fully inhibiting the associated ˙OH production. Two of them, namely the IIcD and IbG analogues, were identified as the ones with the best multifunctional AOC. They both fulfill the Lipinski's and Ghose's rules for orally active drugs. However, IIcD has been chosen as the best prospect for possible application based on potential toxicity and synthetic accessibility estimations. Hopefully, these results might provide motivation for further investigations on the subject, and the synthesis of this compound, so its potential role as protector against oxidative stress – and the associated health issues – could be experimentally confirmed or refuted.
 Please wait while we load your content...
Please wait while we load your content...

