
Reverse regioselectivity in Pd(0)/InI-mediated allylation of aldehydes with ε-amido-allylindiums generated from β-lactams. A new entry to non-racemic highly substituted γ-butyrolactones†
Urszula Klimczak,
Olga Staszewska-Krajewska and
Bartosz K. Zambroń*
Institute of Organic Chemistry, Polish Academy of Sciences, Kasprzaka 44/52, 01-224 Warsaw, Poland. E-mail: bartosz.zambron@icho.edu.pl
First published on 2nd March 2016
Abstract
ε-Amido-allylindiums generated from N-Ts-β-lactams in the presence of 2 eq. of InI, catalytic amounts of Pd(PPh3)4 and 5 eq. of CF3CO2H react regio- and stereoselectively with a number of aromatic and aliphatic aldehydes to afford γ-butyrolactones as the exclusive products in high yield.
Introduction
Due to the well-known antibiotic activity of β-lactams (azetidin-2-ones) a great number of methods for their asymmetric synthesis have been developed.1 Thus a variety of them are readily available in both enantiomeric forms in excellent optical purity. The high reactivity of β-lactams resulting from the strained 4-membered ring, the chirality content that can be easily transferred into a variety of products and the rigidity of their structure, which often makes the reactions involving them very stereoselective make azetidin-2-ones a very interesting chiral building blocks potentially useful in the synthesis of a variety of derivatives not containing a β-lactam fragment. Although there is a significant body of literature on the synthesis of β-lactams, fewer examples of the use of these compounds as synthons in organic synthesis have been reported,2 including an example of their umpolung by Pd(0)/InI methodology to enable the reaction with aldehydes to give (3Z)-2,6-anti-enediols regio- and stereoselectively, developed by our group (Scheme 1).3 During this investigation we have found that performing the reaction in MeOH, in contrast to reactions carried out in aprotic solvents, gave rise to a complex mixture of products among which, after careful examination, slight amounts of competitive γ-addition products were detected. We realized that these compounds must have arisen from the reaction of linear allylindium 3 formed from the initially generated ε-amido-allylindium 2 by chemoselective protonation of N-Ts amido group by solvent molecules (Scheme 2). Indeed, further study confirmed that the regioselectivity of the allylation of aldehydes with ε-amido-allylindiums can be effectively reversed by the addition of a proton source, providing 1,3-diols readily convertible into synthetically and biologically important trisubstituted γ-butyrolactones4 regio- and stereoselectively, which is exemplified in the following article. | ||
| Scheme 1 Reactions of 4-vinyl-β-lactams with aldehydes in the presence of InI and catalytic amounts of Pd(0). | ||
 | ||
| Scheme 2 Possible intermediates involved in Pd(0)/InI mediated allylations of aldehydes with 4-vinyl-β-lactams. | ||
Results and discussion
As a model β-lactam, racemic N-Ts-4-vinylazetidin-2-one 1, which proved to be very resultful in our previous studies, was chosen.3 Isobutyric aldehyde was used as a model electrophile. All initially attempted Barbier-type allylations using other common protic solvents (EtOH, i-PrOH, AcOH) or aprotic solvents of different types and polarities (DCM, THF, DME, dioxane, DMF, NMP, CH3CN) with the addition of protic acids (H2O, AcOH, CF3CO2H, PTSA, TfOH) failed. In all these cases mixtures of α- and γ-adducts with the predominance of the former, accompanied by different amounts of side products including N-Ts amides 4 were obtained. Importantly, within this investigation it turned out that the initially formed γ-adducts (1,3-diols) are unstable and on standing transform slowly into the corresponding γ-butyrolactones via intramolecular TsNH2 substitution. Further study revealed that this process can be effectively accelerated by heating in the presence of DMAP as a nucleophilic catalyst. Therefore in the further investigation all initially formed 1,3-diols were converted without isolation into stable γ-butyrolactones, which were then purified and fully characterised.Suspecting the unprotonated allylindium 2 to be responsible for the presence of linear α-adducts (2,6-enediols), we decided to separate the steps of generation and addition of 3 to the aldehyde to avoid this undesired reaction. A series of experiments with the use of previously mentioned solvents and protic acids as additives let us establish that the addition of 5 eq. of CF3CO2H to the 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 THF:HMPA mixture is superior to other conditions. Using this method, 2 of the 4 possible γ-butyrolactones were obtained in 75% total yield and with moderate 69:31 diastereoselectivity accompanied by only 7% of reduction products 4. The addition of 1 eq. of CF3CO2H led to the formation of linear α-adducts in great predominance and more than 5 eq. had no significant influence on product distribution. The use of PTSA and TfOH resulted in a significant increase in the amount of side products, including 4. The application of H2O and AcOH led, in turn, to the predominance of undesired α-adducts. The use of other solvents, especially polar ones (DMF, NMP) and attempts of replacing HMPA with less toxic DMPU caused a significant increase in the amount of reduction products 4.
:1 THF:HMPA mixture is superior to other conditions. Using this method, 2 of the 4 possible γ-butyrolactones were obtained in 75% total yield and with moderate 69:31 diastereoselectivity accompanied by only 7% of reduction products 4. The addition of 1 eq. of CF3CO2H led to the formation of linear α-adducts in great predominance and more than 5 eq. had no significant influence on product distribution. The use of PTSA and TfOH resulted in a significant increase in the amount of side products, including 4. The application of H2O and AcOH led, in turn, to the predominance of undesired α-adducts. The use of other solvents, especially polar ones (DMF, NMP) and attempts of replacing HMPA with less toxic DMPU caused a significant increase in the amount of reduction products 4.
Since the progress of the reaction is difficult to follow by TLC, to check the reaction completion a 1H NMR study of the crude reaction mixtures quenched after different reaction times was carried out, which led to a very interesting observation (Table 1). Surprisingly, in the reaction mixtures quenched after 30 min and 1 h (Table 1, entries 1 and 2) besides the expected branched 1,3-diols (γ-adducts) and increased amounts of reduction products 4, indicating incomplete conversion, significant amounts of (Z)- and (E)-enediols 5(Z) and 5(E) resulting from α-addition were detected (Fig. 1). The absence of these products in the reaction mixtures quenched after 12 h and 24 h (Table 1, entries 3 and 4), while the total yield of all products was maintained, indicates reversibility of the process. Although reversible allylations5,6 including an example of allylindium(III) species generated in the presence of Pd(0) and InI5b have been reported, to the best of our knowledge this is the first time when the branched homoallylic alcohols resulting from γ-addition are the thermodynamically favoured products (as opposed to the less-hindered linear ones). This observation was further confirmed by low temperature experiments (Table 1, entries 5–8) especially by the case of the reaction conducted in −78 °C, which resulted in the formation of a mixture of γ-butyrolactones 5a, 5b and (Z)- and (E)-enediols 5a(Z), 5b(E) in almost 1:1 ratio (Table 1, entry 7). When the thus formed reaction mixture was removed from the cooling bath and left to stand at 25 °C, within 12 h all initially formed α-adducts 5(Z), 5(E) isomerised completely into branched γ-adducts, which were then further converted into butyrolactones 5a, 5b with comparable yield and diastereoselectivity (Table 1, entry 8). Importantly, when the order of addition of the reagents was partially reversed and CF3CO2H was added to the unquenched reaction mixture containing exclusively α-adducts 5(Z), 5(E) formed via the reaction of allylindium 2 generated in the absence of a proton source, the isomerisation did not occur at all, and no traces of branched γ-adducts (1,3-diols) were detected within the next 12 h (Scheme 3). This outcome stays in line with the results of the initially attempted Barbier-type allylations and shows that only α-allylations involving protonated allylindium 3 are reversible. This confirms that separating the steps of the formation of 3 and its addition to the aldehyde is crucial.
|
|||||
|---|---|---|---|---|---|
| Entry | T [°C] | Time [h] | 5a + 5b yielda [%]; (d.r.)b | 5(Z) + 5(E) yielda [%]; (d.r.)b | 4 yieldc [%] |
| a Isolated yields.b Assayed by 1H NMR integration.c Assayed by crude 1H NMR integration.d After 12 h at −78 °C the cooling bath was removed and the reaction mixture was stirred at 25 °C for additional 12 h. | |||||
| 1 | 25 | 0.5 | 47 (71:29) |
12 (8:92) |
19 |
| 2 | 25 | 1 | 62 (71:29) |
6 (4:96) |
15 |
| 3 | 25 | 12 | 75 (69:31) |
— | 7 |
| 4 | 25 | 24 | 72 (68:32) |
— | 7 |
| 5 | 0 | 12 | 74 (70:30) |
— | 6 |
| 6 | −25 | 12 | 67 (72:28) |
Traces | 5 |
| 7 | −78 | 12 | 27 (73:27) |
31 (15:75) |
16 |
| 8 | −78d | 12 | 77 (72:28) |
— | 6 |

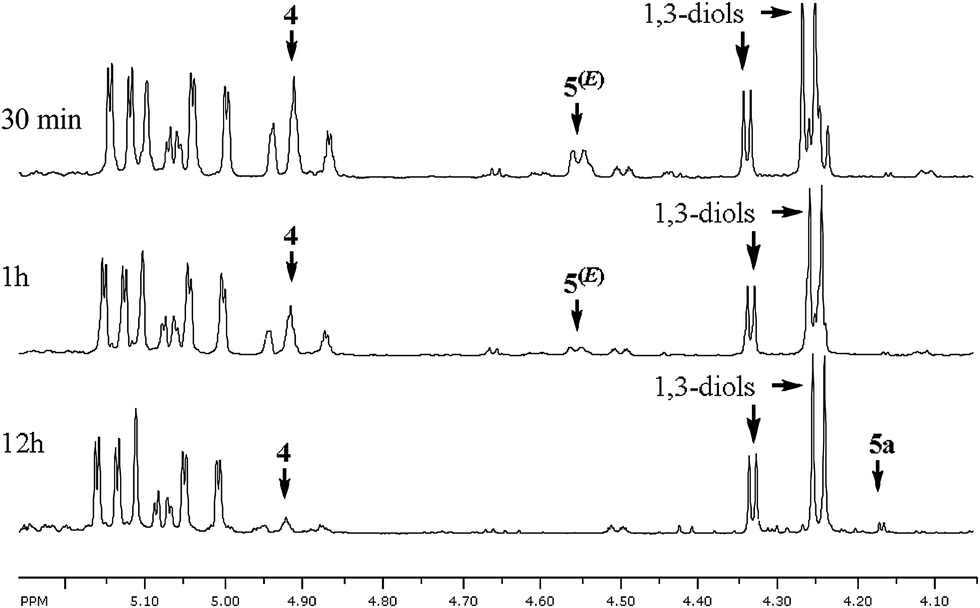 | ||
| Fig. 1 1H NMR study of Pd(PPh3)4/InI-mediated addition of N-Ts-4-vinylazetidin-2-one 1 to isobutyric aldehyde. | ||
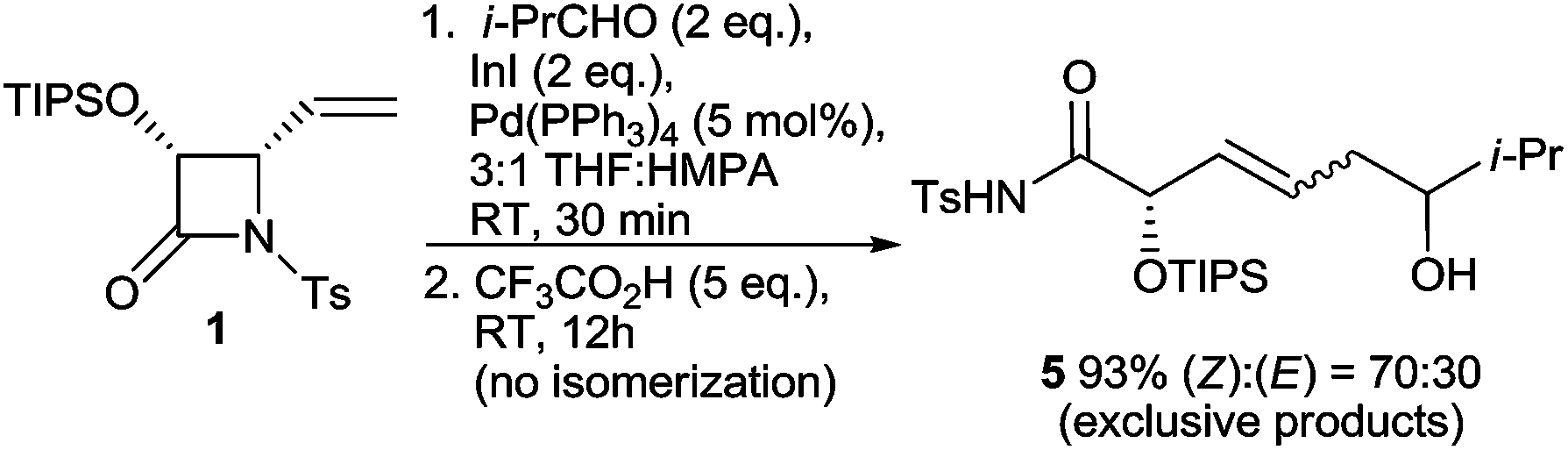 | ||
| Scheme 3 Attempt of isomerizing of the α-dducts 5(E) and 5(Z) (formed in situ via addition of allylindium 2 to isobutyric aldehyde) into its γ-isomers 5 by addition of 5 eq. of CF3CO2H. | ||
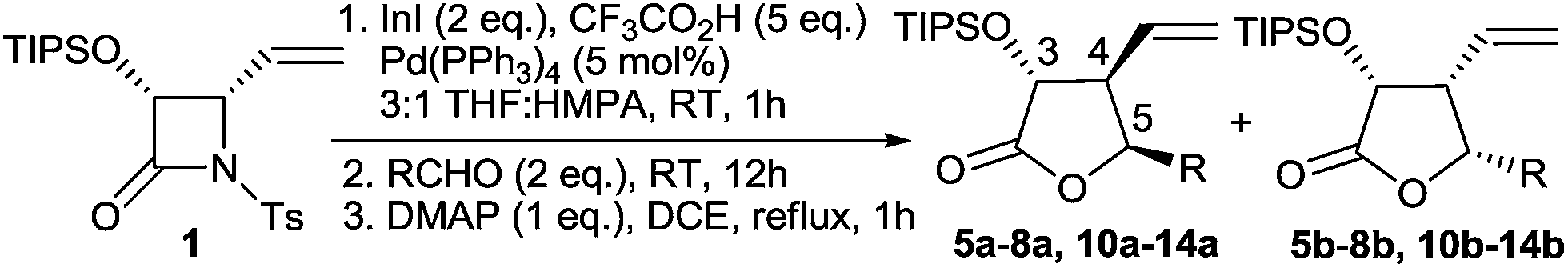
To further explore the scope of the reaction, a variety of aliphatic and aromatic aldehydes were subjected to the optimized reaction conditions. The conducted experiments revealed high sensitivity of the process to the aldehyde type and substitution pattern (Table 2). The reaction of another secondary aliphatic aldehyde, cyclopentanecarboxaldehyde, proceeded with almost the same yield (75%) and diastereoselectivity (71:29, Table 2, entry 2). However, the use of less-hindered primary aldehydes caused a serious decrease of stereoselectivity and, in effect, mixtures of all four possible diastereoisomers were obtained (Table 2, entries 3 and 4). Using pivalaldehyde, in turn, afforded 2,6-enediols 9(Z) and 9(E) in 36% total yield accompanied by significant amounts of 4 instead of the expected γ-butyrolactones. This is most likely due to the severe steric hindrance of the t-Bu substituent preventing the reaction at the more hindered γ-position of the allyl system (Table 2, entry 5). All reactions involving aromatic aldehydes, either electron-rich or electron-deficient, afforded γ-butyrolactones in a significantly higher yield and with better diastereoselectivity (>80:20). However, in all these cases 3,4-cis-4,5-cis-isomers were obtained as the major ones instead of 3,4-trans-4,5-cis-isomers (Table 2, entries 6–9), which was assigned by NOE measurements and confirmed by X-ray analysis of 3,4-cis-4,5-cis-10b (Fig. 2). Finally, employing conjugated 3-methyl-2-butenal also gave the two expected isomers, but with 13b as the major product, analogously to the reactions involving aromatic aldehydes (Table 2, entry 10).
|
||||
|---|---|---|---|---|
| Entry | R | a; yielda [%] | b; yielda [%] | Others; yielda [%] |
| a Isolated yields.b Compounds 7c and 8c are 3,4-cis-4,5-trans substituted and 7d and 8d are 3,4-trans-4,5-trans substituted.c Assayed by 1H NMR integration of an inseparable mixtures of 8c + 8d and 9(Z) + 9(E).d Containing 11% of another diastereoisomer according to 1H NMR.e Assayed by crude 1H NMR integration (2 diastereoisomers in 1:1 ratio). |
||||
| 1 | i-Pr | 5a 52 | 5b 23 | Traces |
| 2 | c-Pent | 6a 53 | 6b 22 | Traces |
| 3 | Et | 7a 23 | 7b 10 | 7cb 38, 7db 8 |
| 4 | PhCH2CH2 | 8a 16 | 8b 11 | 8cb 42c, 8db 10c |
| 5 | t-Bu | — | — | 9(Z) 7c, 9(E) 29c |
| 6 | Ph | 10a 14 | 10b 64 | Traces |
| 7 | p-CN-Ph | 11a 15d | 11b 66 | Traces |
| 8 | p-OMe-Ph | 12a 12 | 12b 62 | Traces |
| 9 |  |
13a 16 | 13b 63 | Traces |
| 10 |  |
14a 14 | 14b 52 | 8e |
 | ||
| Fig. 2 X-ray structure of 10b. | ||
Next, a series of experiments using modified azetidin-2-ones were conducted to determine the effect of β-lactam structure on the reaction outcome (Table 3). As in our previous study,3 attempts of replacing the Ts group with common Boc and PMP protective groups failed. In these cases no conversion was observed, confirming that a strongly electron withdrawing group attached to the nitrogen atom of the β-lactam is necessary. On the other hand, treatment of azetidin-2-one 15 containing the mesyl activating group with benzaldehyde and butyric aldehyde under the conditions developed gave the expected products in somewhat higher total yields (>80%) and with improved selectivity (Table 3, entries 1 and 2). The reaction with benzaldehyde proved to be especially successful, since γ-butyrolactones 10a–10b were obtained in 81% total yield and with excellent 95:5 diastereoselectivity. The reaction of azetidin-2-one 16 containing an i-Pr substituent on C3 of the β-lactam ring, in turn, afforded γ-butyrolactones 17a–17b and 18a–18b in good total yields, although with reduced diastereoselectivity, showing that the reaction is also highly sensitive to the β-lactam substitution (Table 3, entries 3 and 4).
|
|||||
|---|---|---|---|---|---|
| Entry | β-Lactam | R′ | a; yielda [%] | b; yielda [%] | Others; yielda [%] |
| a Isolated yields.b The cyclisation was run in a sealed tube using DCM instead of DCE due to high volatility of the products.c Assayed by crude 1H NMR (single diastereoisomer).d Compound 18c is 3,4-trans-4,5-trans substituted. | |||||
| 1 | 15 | i-Pr | 5a 60 | 5b 23 | Traces |
| 2 | 15 | Ph | 10a 4 | 10b 77 | Traces |
| 3b | 16 | i-Pr | 17a 33 | 17b 31 | 5c |
| 4 | 16 | Ph | 18a 16 | 18b 50 | 18c 14d |

To verify if the method may be applied in asymmetric synthesis, enantioenriched 4-vinylazetidin-2-one (−)-19 (>99% ee according to HPLC, see ESI†),3,7 was treated with isobutyric aldehyde and benzaldehyde under optimized reaction conditions (Scheme 4). As a result of these experiments, the expected γ-butyrolactones (−)-3,4-trans-4,5-cis-5a and (−)-3,4-cis-4,5-cis-10b were obtained as the major products in good yield and with high enantiomeric excess, which was proven by 19F NMR study of the corresponding Mosher's esters 19, 20.
 | ||
| Scheme 4 Pd(PPh3)4/InI-mediated addition of enantioenriched β-lactam (−)-1 to aldehydes. | ||
Finally, to gain insight into the possible mechanism of the process, several additional experiments were carried out. The introduction of isolated (Z)- or (E)-enediols 5(Z), 5b(E) into the unquenched reaction mixture, containing the thermodynamically favoured γ-adducts 5a and 5b exclusively, showed that only indium alkoxides of enediols 5(Z) and 5(E) formed in the course of the reaction can isomerise into their branched γ-isomers under the conditions developed as no change in product distribution was observed within 12 h (Scheme 5). This result strongly suggests the occurrence of a retroallylation mechanism5 in the course of the process being investigated and not the alternative one based on [3,3]-sigmatropic rearrangement,6 both of which are possible and well described. Moreover, subjecting 5(Z), 5(E) to the typical oxonia-Cope rearrangement conditions6 using both Brønsted and Lewis acids as the catalysts gave no trace of the branched 1,3-diols, which further confirms the above conclusion (Scheme 6). Based on these observations and the regio- and stereoselectivity of the process a plausible reaction pathway is depicted in Scheme 7. The initial step consists of C4–N β-lactam bond cleavage by Pd(PPh3)4 followed by reductive transmetalation of the transient π-allylpalladium(II) complex with InI and subsequent chemoselective protonation of the N-Ts-amido group present in the molecule. Within this sequence, typically fast isomerisation on the carbon atom attached to the metal occurs. As a result, linear ε-amido-allylindium 21a existing in equilibrium with its cyclic isomer 21b is generated. Simultaneous reactions of these two species in the following addition step lead to the initial formation of alkoxides 22 and 23 resulting from reactions on the α- or γ-position of the allyl system. However, due to stabilisation of cyclic allylindium 21b by intramolecular coordination of the amido group to the extraordinarily oxophilic indium(III) atom8 and clearly less efficient effect of this type in the linear (Z)- and (E)- adducts 22, the retroallylation process is accelerated and the addition of 21b to the aldehyde is reversible. Conversely, the more efficient stabilisation of the cyclic adduct 23 as compared to the initial linear allylindium 21a by an analogous effect makes the reaction irreversible in this case and results in the preferential formation of branched homoallylic alcohols 24 on prolonged reaction time. Analogously to the original proposal by Takemoto et al. for similar reactions of 2-vinylaziridines,9 addition of 21a to the aldehyde occurs via the most favourable chair-like transition states ts1 and ts2, where R or R′′ is favourably located in the anti-orientation according to the Felkin-Ahn model, depending on the β-lactam and aldehyde type used. Subsequent protonation and cyclisation via TsNH2 substitution leads to 3,4-trans-4,5-cis-25 and 3,4-cis-4,5-cis-25 products in different ratios and usually with a useful level of diastereoselectivity.
 | ||
| Scheme 5 Attempts of isomerizing of isolated 5(E) and/or 5(Z) under the conditions developed. | ||
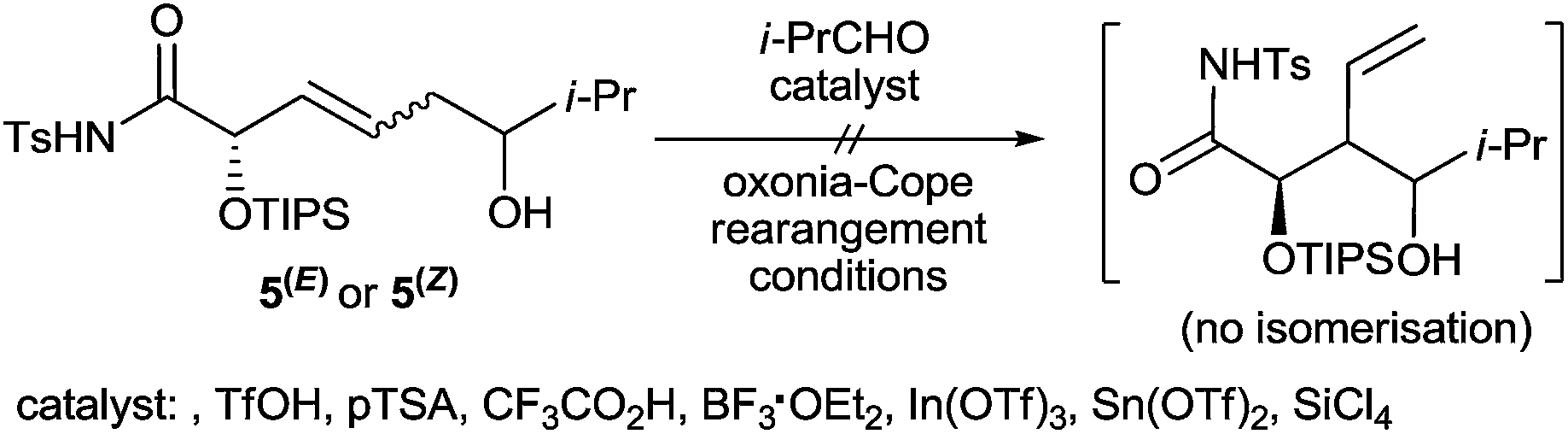 | ||
| Scheme 6 Attempts of isomerizing of isolated 5(E) and/or 5(Z) into 5a and/or 5b under 2-oxonia-Cope rearrangement conditions. | ||
 | ||
| Scheme 7 Possible reaction pathway. | ||
Experimental
Materials and methods
Reagents were purchased from ABCR, Acros, Alfa Aesar or Sigma Aldrich and used as received. InI was powdered in a mortar prior to use. Dry solvents were obtained by distillation over Na/benzophenone (THF) or CaH2 (CH2Cl2). Air- and moisture-sensitive reactions were conducted in oven-dried glassware under argon. Column chromatography was carried out using Kiesel gel (230–400 mesh). Analytical TLC was performed on Silica gel 60 F254 aluminium plates (Merck, Darmstadt). Indication was achieved with UV light (λ = 254 nm) and common dip stains (potassium permanganate or cerium ammonium molybdate). NMR spectra were recorded on Varian Mercury 400 MHz, Bruker DRX 500 MHz and Varian VNMRS 600 MHz spectrometers in CDCl3 or C6D6 solutions (unless indicated otherwise). Chemical shifts are quoted on the δ scale, ppm, with the solvent signal as the internal standard (1H NMR: CDCl3: 7.26 ppm, C6D6: 7.16 ppm, CD3OD: 3.31 ppm; 13C NMR: CDCl3: 77.16 ppm, C6D6: 128.06 ppm, CD3OD: 49.00 ppm). Multiplicities for 1H NMR signals are described using the following abbreviations: s = singlet, d = doublet, t = triplet, q = quartet, sep = septet, m = multiplet. Coupling constants, J, are given in Hertz (Hz). Infrared spectra (IR) were measured on a FT-IR-1600-Perkin Elmer spectrophotometer and are reported in cm−1. The samples were prepared as thin films. High resolution mass spectra (HRMS) were obtained on ESI-TOF Mariner spectrometer (Perspective Biosystem) and are given in m/z. Melting points (mp) were determined with Melting Point Meter MPM-H2 apparatus and are uncorrected. Optical rotations [α] were measured on Jasco P-2000 Polarimeter in a quartz glass cuvette at λ = 589 nm (Na D-line). Concentrations [c] are given in g/100 mL. Analytical high performance liquid chromatography (HPLC) was performed using Hitachi/Merck HPLC system (Hitachi L-2130 pump, Diode Array L2450 detector [monitoring at 212 nm]) outfitted with Daicel Chiralpak IB (250 mm × 4.6 mm, 5 μm) column. Operating procedures and retention times are reported with the corresponding chromatograms. The relative stereochemistry of all obtained γ-butyrolactones was assigned by NOE measurements (Varian VNMRS 600 MHz). β-Lactams 1,3,7 (−)-1,3,7 163 and (±)-3-cis-((triisopropylsilyl)oxy)-4-vinylazetidin-2-one3 were prepared following literature procedures and their analytical data were consistent with those published in the literature. The spectral data of α-adducts 9(E) and 9(Z) were in accordance with the reported values.3Synthetic procedures
:1; 2 mL), CF3COOH (41.25 μL, 0.590 mmol; 5 eq.) and a mixture of InI (powdered; 57 mg, 0.236 mmol; 2 eq.) and Pd(PPh3)4 (6.9 mg, 0.006 mmol; 5 mol%) were sequentially added at 25 °C under an atmosphere of argon. After 1 hour, the aldehyde (0.236 mmol; 2 eq.) was added in one portion at the same temperature and stirring was continued for 12 hours. At the end of this time, the mixture was treated with 1 M HCl (2 mL), poured into water (25 mL) and diluted with ethyl acetate (10 mL). The phases were separated and the aqueous one was extracted with an additional portion of EtOAc (10 mL). The combined organic layers were successively washed with NaHCO3 (25 mL), water (2 × 25 mL), brine (25 mL), then dried over MgSO4 and evaporated. The residue was dissolved in dry DME (10 mL), DMAP (14.4 mg, 0.118 mmol; 1 eq.) was added and the mixture was refluxed for 1 hour under an atmosphere of argon. After being cooled to 25 °C, the crude solution was combined with a portion of silica gel and the solvent was removed under reduced pressure. Column chromatography using a diethyl ether/hexane or ethyl acetate/hexane mixture (unless indicated otherwise) enabled the separation of diastereomers.
(3R*,4S*,5R*)-5-Isopropyl-3-((triisopropylsilyl)oxy)-4-vinyldihydrofuran-2(3H)-one (5a). Yield: 20.0 mg (52%); colorless oil; Rf (5% Et2O/hexane) 0.60; 1H NMR (600 MHz, CDCl3) δ: 5.65 (dt, J = 17.0, 10.3, 1H), 5.28–5.19 (m, 2H), 4.31 (dd, J = 9.7, 5.1 Hz, 1H), 4.17 (d, J = 2.2 Hz, 1H), 2.92 (ddd, J = 10.1, 5.1, 2.2 Hz, 1H), 1.85 (dsep, J = 9.7, 6.7 Hz, 1H), 1.19–1.11 (m, 3H), 1.10–1.03 (m, 21H), 0.87 (d, J = 6.7 Hz, 3H); 13C NMR (151 MHz, CDCl3) δ: 174.7, 131.0, 119.8, 86.9, 75.4, 52.8, 28.7, 19.6, 17.8, 17.7, 17.4, 12.0; IR (film) ν: 3083, 2944, 2868, 1787, 1642, 1467, 1390, 1177, 1110, 988, 882, 683 cm−1; HRMS (ESI-TOF) m/z calcd for C18H34O3SiNa [M + Na+] 349.2175. Found 349.2181.
(3S,4R,5S)-5-Isopropyl-3-((triisopropylsilyl)oxy)-4-vinyldihydrofuran-2(3H)-one ((−)-5a). Yield: 19.3 mg (50%); ee > 99% (19F NMR analysis of Mosher's ester 19); colorless oil; Rf (5% Et2O/hexane) 0.60; [α]23D = −41.1 (c 0.75, CHCl3); NMR and IR spectra were consistent with those recorded for 5a; HRMS (ESI-TOF) m/z calcd for C18H34O3SiNa [M + Na+] 349.2175. Found 349.2173.
(3S*,4S*,5R*)-5-Isopropyl-3-((triisopropylsilyl)oxy)-4-vinyldihydrofuran-2(3H)-one (5b). Yield: 8.9 mg (23%); colorless oil Rf (5% Et2O/hexane) 0.45; 1H NMR (600 MHz, CDCl3) δ: 5.63 (ddd, J = 17.0, 10.7, 10.4 Hz, 1H), 5.28 (dd, J = 10.4, 1.4 Hz, 1H), 5.20 (dd, J = 17.0, 1.4 Hz, 1H), 4.64 (d, J = 6.9 Hz, 1H), 3.79 (dd, J = 10.5, 4.1 Hz, 1H), 3.16 (ddd, J = 10.7, 6.9, 4.1 Hz, 1H), 1.82 (dsep, J = 10.5, 6.6 Hz, 1H), 1.16–1.10 (m, 3H), 1.09–1.04 (m, 21H), 0.84 (d, J = 6.6 Hz, 3H); 13C NMR (151 MHz, CDCl3) δ: 175.1, 129.9, 120.5, 83.4, 72.3, 51.0, 29.0, 19.6, 17.8, 17.7, 16.7, 12.0; IR (film) ν: 3080, 2944, 2868, 1789, 1645, 1466, 1390, 1167, 1057, 989, 882, 685 cm−1; HRMS (ESI-TOF) m/z calcd for C18H34O3SiNa [M + Na+] 349.2175. Found 349.2182.
(3R*,4S*,5R*)-5-Cyclopentyl-3-((triisopropylsilyl)oxy)-4-vinyldihydrofuran-2(3H)-one (6a). Yield: 22.1 mg (53%); colorless oil; Rf (5% Et2O/hexane) 0.60; 1H NMR (600 MHz, CDCl3) δ: 5.68 (dt, J = 16.9, 10.2 Hz, 1H), 5.25–5.20 (m, 2H), 4.46 (dd, J = 9.5, 5.7 Hz, 1H), 4.21 (d, J = 3.4 Hz, 1H), 2.95–2.89 (m, 1H), 2.08–1.99 (m, 1H), 1.91–1.84 (m, 1H), 1.75–1.68 (m, 1H), 1.68–1.59 (m, 2H), 1.58–1.50 (m, 2H), 1.45–1.37 (m, 1H), 1.21–1.10 (m, 4H), 1.10–1.04 (m, 18H); 13C NMR (151 MHz, CDCl3) δ: 174.8, 131.9, 119.6, 85.6, 74.7, 53.3, 40.4, 30.3, 27.8, 25.5, 25.1, 17.8, 17.7, 12.1; IR (film) ν: 3081, 2945, 2867, 1785, 1643, 1464, 1152, 1107, 882, 683 cm−1; HRMS (ESI-TOF) m/z calcd for C20H36O3SiNa [M + Na+] 375.2331. Found 375.2328.
(3S*,4S*,5R*)-5-Cyclopentyl-3-((triisopropylsilyl)oxy)-4-vinyldihydrofuran-2(3H)-one (6b). Yield: 9.2 mg (22%); colorless oil; Rf (5% Et2O/hexane) 0.45; 1H NMR (600 MHz, CDCl3) δ: 5.68 (ddd, J = 16.9, 10.8, 10.4 Hz, 1H), 5.28 (dd, J = 10.4, 1.4 Hz, 1H), 5.18 (dd, J = 16.9, 1.4 Hz, 1H), 4.64 (d, J = 6.9 Hz, 1H), 3.98 (dd, J = 10.5, 4.3 Hz, 1H), 3.11 (ddd, J = 10.8, 6.9, 4.3 Hz, 1H), 2.07–1.99 (m, 1H), 1.97–1.90 (m, 1H), 1.76–1.69 (m, 1H), 1.64–1.57 (m, 2H), 1.57–1.50 (m, 2H), 1.44–1.37 (m, 1H), 1.19–1.10 (m, 3H), 1.09–1.05 (m, 19H); 13C NMR (151 MHz, CDCl3) δ: 175.3, 130.6, 120.3, 83.1, 72.1, 51.5, 40.5, 30.6, 27.1, 25.3, 25.1, 17.8, 17.7, 12.0; IR (film) ν: 3079, 2945, 2867, 1788, 1645, 1464, 1162, 992, 882, 685 cm−1; HRMS (ESI-TOF) m/z calcd for C20H36O3SiNa [M + Na+] 375.2331. Found 375.2330.
5-Ethyl-3-((triisopropylsilyl)oxy)-4-vinyldihydrofuran-2(3H)-ones (7). Crude products were chromatographed on silica gel using MTBE/c-hexane to provide pure samples of 7a and 7b and a mixture of 7c and 7d. The latter compounds were separated by further column chromatography eluting with Et2O/n-pentane.
(3R*,4S*,5R*)-5-Ethyl-3-((triisopropylsilyl)oxy)-4-vinyldihydrofuran-2(3H)-one (7a). Yield: 8.5 mg (23%); colorless oil; Rf (5% MTBE/c-hexane) 0.65; 1H NMR (600 MHz, CDCl3) δ: 5.72–5.64 (m, 1H), 5.29–5.22 (m, 2H), 4.53 (dt, J = 8.1, 6.3 Hz, 1H), 4.30 (d, J = 5.7 Hz, 1H), 3.05–2.98 (m, 1H), 1.63–1.58 (m, 2H), 1.19–1.11 (m, 3H), 1.10–1.04 (m, 18H), 1.01 (t, J = 7.4 Hz, 3H); 13C NMR (151 MHz, CDCl3) δ: 174.7, 131.9, 120.1, 82.0, 73.7, 53.1, 24.3, 17.84, 17.78, 12.2, 10.1; IR (film) ν: 3086, 2943, 2867, 1789, 1643, 1464, 1152, 1105, 882, 684 cm−1; HRMS (ESI-TOF) m/z calcd for C17H32O3SiNa [M + Na+] 335.2018. Found 335.2017.
(3S*,4S*,5R*)-5-Ethyl-3-((triisopropylsilyl)oxy)-4-vinyldihydrofuran-2(3H)-one (7b). Yield: 3.7 mg (10%); colorless oil; Rf (5% MTBE/c-hexane) 0.50; 1H NMR (600 MHz, CDCl3) δ: 5.68 (dt, J = 17.0, 10.3 Hz, 1H), 5.28 (dd, J = 10.3, 1.3 Hz, 1H), 5.19 (dd, J = 17.0, 1.3 Hz, 1H), 4.63 (d, J = 6.9 Hz, 1H), 4.21 (ddd, J = 8.0, 6.1, 4.9 Hz, 1H), 3.13 (m, 1H), 1.78–1.68 (m, 1H), 1.57–1.49 (m, 1H), 1.18–1.11 (m, 3H), 1.09–1.05 (m, 18H), 0.97 (t, J = 7.4 Hz, 3H); 13C NMR (151 MHz, CDCl3) δ: 175.1, 130.2, 120.4, 80.0, 71.9, 51.2, 24.4, 17.8, 17.7, 12.1, 9.5; IR (film) ν: 3080, 2944, 2867, 1790, 1645, 1465, 1170, 1135, 992, 885, 686 cm−1; HRMS (ESI-TOF) m/z calcd for C17H32O3SiNa [M + Na+] 335.2018. Found 335.2014.
(3R*,4R*,5R*)-5-Ethyl-3-((triisopropylsilyl)oxy)-4-vinyldihydrofuran-2(3H)-one (7c). Yield: 14.0 mg (38%); colorless oil; Rf (5% Et2O/n-pentane) 0.75; 1H NMR (600 MHz, CDCl3
:C6D6 3:2) δ: 6.02 (ddd, J = 17.3, 10.4, 9.1 Hz, 1H), 5.35 (dd, J = 10.4, 1.8 Hz, 1H), 5.30 (dd, J = 17.3, 1.8 Hz, 1H), 4.49 (d, J = 5.7 Hz, 1H), 4.46 (td, J = 8.1, 3.9 Hz, 1H), 2.69 (ddd, J = 9.1, 8.1, 5.7 Hz, 1H), 1.85 (dqd, J = 15.0, 7.5, 3.9 Hz, 1H), 1.70–1.61 (m, 1H), 1.33–1.25 (m, 3H), 1.21–1.18 (m, 18H), 1.13 (t, J = 7.4 Hz, 3H); 13C NMR (151 MHz, CDCl3:C6D6 3:2) δ: 174.6, 132.1, 119.7, 83.9, 72.4, 52.7, 26.2, 18.0, 17.9, 12.4, 10.0; IR (film) ν: 3082, 2943, 2868, 1788, 1642, 1464, 1140, 1109, 882, 686 cm−1; HRMS (ESI-TOF) m/z calcd for C17H32O3SiNa [M + Na+] 335.2018. Found 335.2017.
(3R*,4S*,5S*)-5-Ethyl-3-((triisopropylsilyl)oxy)-4-vinyldihydrofuran-2(3H)-one (7d). Yield: 3.0 mg (8%); colorless oil; Rf (5% Et2O/n-pentane) 0.70; 1H NMR (600 MHz, CDCl3) δ: 5.71 (ddd, J = 17.1, 10.1, 8.9 Hz, 1H), 5.31–5.25 (m, 2H), 4.40 (d, J = 10.0 Hz, 1H), 3.99 (ddd, J = 9.7, 8.0, 3.7 Hz, 1H), 2.74–2.67 (m, 1H), 1.79 (dqd, J = 15.0, 7.5, 3.7 Hz, 1H), 1.67–1.60 (m, 1H), 1.19–1.11 (m, 3H), 1.09–1.06 (m, 18H), 1.03 (t, J = 7.5 Hz, 3H); 13C NMR (151 MHz, CDCl3) δ: 174.6, 134.3, 120.4, 80.3, 74.6, 55.7, 26.2, 17.90, 17.87, 12.4, 9.7; IR (film) ν: 3083, 2943, 2868, 1791, 1646, 1464, 1165, 883, 683 cm−1; HRMS (ESI-TOF) m/z calcd C17H32O3SiNa [M + Na+] 335.2018. Found 335.2018.
5-Phenethyl-3-((triisopropylsilyl)oxy)-4-vinyldihydrofuran-2(3H)-ones (8). Column chromatography of crude products eluting with EtOAc/hexane provided pure compound 8a. Lactones 8b and 8c were inseparable and thus characterized as a mixture. Diastereomer 8d was isolated after the removal of excess aldehyde by further column chromatography using Et2O/n-pentane.
(3R*,4S*,5R*)-5-Phenethyl-3-((triisopropylsilyl)oxy)-4-vinyldihydrofuran-2(3H)-one (8a). Yield: 7.3 mg (16%); colorless oil; Rf (10% ethyl acetate/hexane) 0.70; 1H NMR (600 MHz, CDCl3) δ: 7.31–7.28 (m, 2H), 7.23–7.18 (m, 3H), 5.69 (dt, J = 16.7, 10.1 Hz, 1H), 5.28–5.23 (m, 2H), 4.66–4.60 (m, 1H), 4.32 (d, J = 5.8 Hz, 1H), 3.05–2.99 (m, 1H), 2.88–2.82 (m, 1H), 2.69 (dt, J = 13.8, 8.1 Hz, 1H), 1.89–1.83 (m, 2H), 1.18–1.12 (m, 3H), 1.10–1.05 (m, 18H); 13C NMR (101 MHz, CDCl3) δ: 174.6, 140.8, 131.9, 128.52, 128.48, 126.2, 120.4, 79.7, 73.7, 53.1, 33.3, 32.0, 17.9, 17.8, 12.2; IR (film) ν: 3085, 3027, 2925, 2866, 1787, 1644, 1604, 1462, 1151, 1013, 882, 685 cm−1; HRMS (ESI-TOF) m/z calcd for C23H36O3SiNa [M + Na+] 411.2331. Found 411.2329.
(3S*,4S*,5R*)-5-Phenethyl-3-((triisopropylsilyl)oxy)-4-vinyldihydrofuran-2(3H)-one (8b). Yield: 5.0 mg (11%); colorless crystals; mp: 50.4–52.3 °C; Rf (10% ethyl acetate/hexane) 0.60; 1H NMR (600 MHz, CDCl3) δ: 7.32–7.28 (m, 2H), 7.23–7.17 (m, 3H), 5.71 (dt, J = 17.0, 10.3 Hz, 1H), 5.30 (dd, J = 10.3, 1.3 Hz, 1H), 5.20 (dd, J = 17.0, 1.3 Hz, 1H), 4.63 (d, J = 7.0 Hz, 1H), 4.29 (dt, J = 9.3, 4.7 Hz, 1H), 3.15–3.08 (m, 1H), 2.85–2.78 (m, 1H), 2.73–2.66 (m, 1H), 2.06–1.98 (m, 1H), 1.81–1.74 (m, 1H), 1.19–1.11 (m, 3H), 1.10–1.05 (m, 18H); 13C NMR (151 MHz, CDCl3) δ: 175.0, 140.7, 130.3, 128.50, 128.49, 126.1, 120.6, 77.4, 71.8, 51.4, 33.2, 31.4, 17.8, 17.7, 12.1; IR (film) ν: 3083, 3027, 2944, 2867, 1790, 1458, 1160, 1007, 883, 686 cm−1; HRMS (ESI-TOF) m/z calcd for C23H36O3SiNa [M + Na+] 411.2331. Found 411.2326.
(3R*,4R*,5R*)-5-Phenethyl-3-((triisopropylsilyl)oxy)-4-vinyldihydrofuran-2(3H)-one (8c) and (3R*,4S*,5S*)-5-phenethyl-3-((triisopropylsilyl)oxy)-4-vinyldihydrofuran-2(3H)-one (8d). Characterized as a mixture. Yield: 23.8 mg (52%; 8c/8d = 4.2/1); colorless oil; Rf (10% ethyl acetate/hexane) 0.65; 1H NMR (400 MHz, C6D6) δ: 7.14–7.10 (m, 2Hb + 2Hc), 7.08–7.02 (m, 1Hb + 1Hc), 7.02–6.97 (m, 2Hb + 2Hc), 5.81–5.68 (m, 1Hb), 5.18–5.07 (m, 1Hc), 4.96 (dd, J = 10.3, 1.4 Hz, 1Hb), 4.92–4.89 (m, 1Hc), 4.85 (m, 1Hb + 1Hc), 4.27 (td, J = 8.7, 3.2 Hz, 1Hb), 4.17 (d, J = 5.6 Hz, 1Hb), 4.03 (d, J = 10.2 Hz, 1Hc), 3.49 (td, J = 9.5, 3.0 Hz, 1Hc), 2.76–2.67 (m, 1Hb), 2.67–2.61 (m, 1Hc), 2.56–2.36 (m, 1Hb + 2Hc), 2.03 (td, J = 8.7, 5.6 Hz, 1Hb), 1.76–1.63 (m, 1Hb + 1Hc), 1.63–1.41 (m, 1Hb + 1Hc), 1.21–1.09 (m, 21Hb + 21Hc); 13C NMR (101 MHz, C6D6) δ: 173.4, 173.3, 141.0, 140.8, 134.4, 132.0, 128.4, 128.3, 126.04, 126.02, 119.6, 119.1, 80.9, 76.7, 74.4, 72.3, 56.2, 52.8, 35.1, 34.7, 32.0, 31.4, 17.9, 17.84, 17.82, 17.7, 12.4, 12.3; IR (film) ν: 3084, 3027, 2944, 2867, 1789, 1643, 1604, 1463, 1209, 1152, 1121, 997, 882, 686 cm−1; HRMS (ESI-TOF) m/z calcd for C23H36O3SiNa [M + Na+] 411.2331. Found 411.2330.
(3R*,4S*,5S*)-5-Phenyl-3-((triisopropylsilyl)oxy)-4-vinyldihydrofuran-2(3H)-one (10a). Yield: 6.0 mg (14%); colorless oil; Rf (10% Et2O/hexane) 0.55; 1H NMR (400 MHz, CDCl3) δ: 7.41–7.29 (m, 3H), 7.18–7.13 (m, 2H), 5.69 (d, J = 7.3 Hz, 1H), 5.21–5.06 (m, 3H), 4.47 (d, J = 7.2 Hz, 1H), 3.36–3.27 (m, 1H), 1.22–1.04 (m, 21H); 13C NMR (101 MHz, CDCl3) δ: 174.8, 135.9, 132.8, 128.5, 128.2, 125.7, 120.2, 80.8, 72.8, 54.4, 17.9, 17.8, 12.3; IR (film) ν: 2944, 2867, 1800, 1462, 1308, 1150, 1135, 1003, 882, 707 cm−1; HRMS (ESI-TOF) m/z calcd for C21H32O3SiNa [M + Na+] 383.2018. Found 383.2019.
(3S*,4S*,5S*)-5-Phenyl-3-((triisopropylsilyl)oxy)-4-vinyldihydrofuran-2(3H)-one (10b). Yield: 27.2 mg (64%); colorless crystals; mp: 73.7–74.5 °C; Rf (10% Et2O/hexane) 0.45; 1H NMR (600 MHz, CDCl3) δ: 7.37–7.31 (m, 2H), 7.29–7.25 (m, 1H), 7.24–7.20 (m, 2H), 5.49 (d, J = 5.3 Hz, 1H), 5.42 (dt, J = 17.0, 10.3, 1H), 5.00 (dd, J = 10.3, 1.4 Hz, 1H), 4.96–4.91 (m, 1H), 4.82 (d, J = 6.8 Hz, 1H), 3.42 (ddd, J = 10.1, 6.8, 5.3 Hz, 1H), 1.21–1.10 (m, 3H), 1.11–1.01 (m, 18H); 13C NMR (101 MHz, CDCl3) δ: 174.9, 135.7, 130.1, 128.3, 128.0, 125.7, 120.7, 79.4, 72.2, 53.3, 17.8, 17.7, 12.1; IR (film) ν: 2944, 2867, 1793, 1463, 1385, 1158, 998, 882, 694 cm−1; HRMS (ESI-TOF) m/z calcd for C21H32O3SiNa [M + Na+] 383.2018. Found 383.2018.
(3S,4S,5S)-5-Phenyl-3-((triisopropylsilyl)oxy)-4-vinyldihydrofuran-2(3H)-one ((−)-10b). Yield: 27.6 mg (65%); ee > 99% (19F NMR analysis of Mosher's ester 20); colorless oil; Rf (10% Et2O/hexane) 0.45; [α]23D = −5.8 (c 1.38, CHCl3); NMR and IR data were consistent with those obtained for 10b; HRMS (ESI-TOF) m/z calcd for C21H32O3SiNa [M + Na+] 383.2018.
(3R*,4S*,5S*)-5-(4-Cyanophenyl)-3-((triisopropylsilyl)oxy)-4-vinyldihydrofuran-2(3H)-one (11a). Obtained as inseparable mixture (6.8 mg, 15%; 11a/11c = 9/1); colorless oil; Rf (20% Et2O/hexane) 0.45; 1H NMR (600 MHz, CDCl3) δ: 7.69–7.65 (m, 2Ha + 2Hc), 7.45–7.40 (m, 2Hc), 7.33–7.30 (m, 2Ha), 6.03 (ddd, J = 17.3, 10.3, 9.0 Hz, 1Hc), 5.79 (d, J = 6.8 Hz, 1Ha), 5.44 (d, J = 8.6 Hz, 1Hc), 5.29 (dd, J = 10.3, 1.0 Hz, 1Hc), 5.19–5.09 (m, 3Ha), 5.04 (dd, J = 17.3, 1.0 Hz, 1Hc), 4.49 (d, J = 5.3 Hz, 1Hc), 4.38 (d, J = 5.3 Hz, 1Ha), 3.35–3.28 (m, 1Ha), 2.76 (td, J = 9.0, 8.6, 5.3 Hz, 1Hc), 1.19–1.13 (m, 3Ha + 3Hc), 1.11–1.05 (m, 18Ha + 18Hc); 13C NMR (151 MHz, CDCl3) δ: 173.9, 141.3, 132.3, 131.4, 126.4, 120.9, 118.3, 112.1, 80.2, 73.3, 54.3, 17.8, 17.7, 12.1; IR (film) ν: 3083, 2944, 2867, 2230, 1803, 1612, 1464, 1150, 1134, 1008, 832, 685 cm−1; HRMS (ESI-TOF) m/z calcd for C22H31NO3SiNa [M + Na+] 408.1971. Found 408.1967.
(3S*,4S*,5S*)-5-(4-Cyanophenyl)-3-((triisopropylsilyl)oxy)-4-vinyldihydrofuran-2(3H)-one (11b). Yield: 30.0 mg (66%); colorless crystals; mp: 92.3–93.4 °C; Rf (20% Et2O/hexane) 0.20; 1H NMR (400 MHz, CDCl3) δ: 7.67–7.60 (m, 2H), 7.40–7.32 (m, 2H), 5.53 (d, J = 5.4 Hz, 1H), 5.34 (ddd, J = 16.9, 10.3, 10.1 Hz, 1H), 5.04 (dd, J = 10.3, 1.3 Hz, 1H), 4.97 (dd, J = 16.9, 1.3 Hz, 1H), 4.83 (d, J = 6.8 Hz, 1H), 3.46 (ddd, J = 10.1, 6.8, 5.4 Hz, 1H), 1.19–1.03 (m, 21H); 13C NMR (101 MHz, CDCl3) δ: 174.2, 141.3, 132.2, 129.5, 126.5, 121.5, 118.4, 112.0, 78.5, 71.9, 52.9, 17.8, 17.7, 12.1; IR (film) ν: 3081, 2944, 2867, 2229, 1798, 1612, 1464, 1207, 1156, 1004, 882, 686 cm−1; HRMS (ESI-TOF) m/z calcd for C22H31NO3SiNa [M + Na+] 408.1971. Found 408.1962.
5-(4-Methoxyphenyl)-3-((triisopropylsilyl)oxy)-4-vinyldihydrofuran-2(3H)-ones (12). Column chromatography of crude products (Et2O/hexane) provided pure compound 12a. Lactone 12b was isolated after the removal of excess aldehyde in the next chromatographic step (toluene/hexane).
(3R*,4S*,5S*)-5-(4-Methoxyphenyl)-3-((triisopropylsilyl)oxy)-4-vinyldihydrofuran-2(3H)-one (12a). Yield: 5.5 mg (12%); colorless oil; Rf (10% Et2O/hexane) 0.30; 1H NMR (600 MHz, C6D6) δ: 6.90–6.83 (m, 2H), 6.68–6.63 (m, 2H), 5.20 (d, J = 7.7 Hz, 1H), 5.08 (ddd, 17.0, 10.1, 9.4 Hz, 1H), 4.93 (dd, J = 17.0, 1.6 Hz, 1H), 4.83 (dd, J = 10.1, 1.6 Hz, 1H), 4.55 (d, J = 8.3 Hz, 1H), 3.23 (s, 3H), 2.98 (ddd, 9.4, 8.3, 7.7 Hz, 1H), 1.20–1.12 (m, 21H); 13C NMR (151 MHz, C6D6) δ: 174.1, 159.5, 133.7, 128.1, 126.9, 119.4, 113.8, 79.9, 72.4, 54.3, 54.1, 17.80, 17.77, 12.3; IR (film) ν: 2943, 2867, 1798, 1614, 1516, 1254, 1150, 1135, 998, 830, 683 cm−1; HRMS (ESI-TOF) m/z calcd for C22H34O4SiNa [M + Na+] 413.2124. Found 413.2120.
(3S*,4S*,5S*)-5-(4-Methoxyphenyl)-3-((triisopropylsilyl)oxy)-4-vinyldihydrofuran-2(3H)-one (12b). Yield: 28.6 mg (62%); colorless crystals; mp: 79.5–80.6 °C; Rf (10% Et2O/hexane) 0.20; 1H NMR (400 MHz, C6D6) δ: 7.03–6.97 (m, 2H), 6.77–6.71 (m, 2H), 5.44 (ddd, J = 17.0, 10.3, 10.1 Hz, 1H), 4.83 (d, J = 5.3 Hz, 1H), 4.78 (dd, J = 10.3, 1.8 Hz, 1H), 4.73 (ddd, J = 17.0, 1.8, 0.6 Hz, 1H), 4.49 (d, J = 7.0 Hz, 1H), 3.25 (d, J = 2.1 Hz, 3H), 2.91 (ddd, J = 10.1, 7.0, 5.3 Hz, 1H), 1.20–1.13 (m, 21H); 13C NMR (101 MHz, C6D6) δ: 174.0, 159.5, 130.9, 128.0, 127.1, 119.8, 113.6, 78.7, 72.1, 54.4, 53.2, 17.74, 17.69, 12.2; IR (film) ν: 2944, 2867, 1793, 1615, 1517, 1464, 1251, 1159, 998, 882, 686 cm−1; HRMS (ESI-TOF) m/z calcd for C22H34O4SiNa [M + Na+] 413.2124. Found 413.2125.
(3R*,4S*,5S*)-5-(Naphthalen-2-yl)-3-((triisopropylsilyl)oxy)-4-vinyldihydrofuran-2(3H)-one (13a). Yield: 7.8 mg (16%); colorless crystals; mp: 58.4–60.1 °C; Rf (25% ethyl acetate/hexane) 0.75; 1H NMR (400 MHz, CDCl3) δ: 7.86–7.81 (m, 3H), 7.69–7.67 (m, 1H), 7.53–7.47 (m, 2H), 7.24–7.19 (m, 1H), 5.87 (d, J = 7.2 Hz, 1H), 5.23–5.16 (m, 2H), 5.09–5.00 (m, 1H), 4.52 (d, J = 6.8 Hz, 1H), 3.42–3.35 (m, 1H), 1.21–1.04 (m, 21H); 13C NMR (101 MHz, CDCl3) δ: 174.8, 133.5, 133.05, 132.99, 132.7, 128.3, 128.1, 127.7, 126.6, 126.4, 124.6, 123.5, 120.3, 81.0, 73.0, 54.5, 17.9, 17.8, 12.3; IR (film) ν: 3057, 2944, 2867, 1800, 1604, 1464, 1310, 1150, 1135, 1011, 818, 684 cm−1; HRMS (ESI-TOF) m/z calcd for C25H34O3SiNa [M + Na+] 433.2175. Found 433.2171.
(3S*,4S*,5S*)-5-(Naphthalen-2-yl)-3-((triisopropylsilyl)oxy)-4-vinyldihydrofuran-2(3H)-one (13b). Yield: 30.5 mg (63%); colorless crystals; mp: 85.9–87.6 °C; Rf (25% ethyl acetate/hexane) 0.70; 1H NMR (600 MHz, CDCl3) δ: 7.85–7.77 (m, 4H), 7.51–7.46 (m, 2H), 7.32–7.28 (m, 1H), 5.66 (d, J = 5.4 Hz, 1H), 5.47 (dt, J = 17.4, 10.1 Hz, 1H), 4.99–4.94 (m, 2H), 4.89 (d, J = 6.8 Hz, 1H), 3.54 (ddd, J = 10.1, 6.8, 5.4 Hz, 1H), 1.23–1.16 (m, 3H), 1.15–1.03 (m, 18H); 13C NMR (101 MHz, CDCl3) δ: 174.9, 133.3, 133.05, 132.97, 130.1, 128.1, 128.0, 127.7, 126.3, 126.2, 124.7, 123.5, 120.8, 79.5, 72.3, 53.2, 17.83, 17.77, 12.1; IR (film) ν: 2921, 2866, 1793, 1648, 1467, 1156, 1002, 789, 686 cm−1; HRMS (ESI-TOF) m/z calcd for C25H34O3SiNa [M + Na+] 433.2175. Found 433.2171.
(3R*,4S*,5R*)-5-(2-Methylprop-1-en-1-yl)-3-((triisopropylsilyl)oxy)-4-vinyldihydrofuran-2(3H)-one (14a). Yield: 5.6 mg (14%); colorless oil; Rf (5% Et2O/hexane) 0.45; 1H NMR (600 MHz, CD3OD) δ: 5.79 (ddd, J = 17.0, 10.1, 9.4 Hz, 1H), 5.36 (dd, J = 9.8, 7.7 Hz, 1H), 5.32–5.27 (m, 2H), 5.20 (dd, J = 10.1, 1.3 Hz, 1H), 4.59 (d, J = 9.1 Hz, 1H), 3.21–3.16 (m, 1H), 1.82 (d, J = 1.2 Hz, 3H), 1.72 (d, J = 1.1 Hz, 3H), 1.23–1.15 (m, 3H), 1.14–1.09 (m, 18H); 13C NMR (151 MHz, CD3OD) δ: 177.2, 141.7, 134.7, 120.7, 120.3, 78.2, 74.2, 55.0, 25.9, 18.42, 18.38, 13.6; IR (film) ν: 2943, 2867, 1792, 1646, 1465, 1151, 1136, 883, 682 cm−1; HRMS (ESI-TOF) m/z calcd for C19H34O3SiNa [M + Na+] 361.2175. Found 361.2174.
(3S*,4S*,5R*)-5-(2-Methylprop-1-en-1-yl)-3-((triisopropylsilyl)oxy)-4-vinyldihydrofuran-2(3H)-one (14b). Yield: 20.8 mg (52%); colorless oil; Rf (5% Et2O/hexane) 0.30; 1H NMR (600 MHz, C6D6) δ: 5.69 (dt, J = 17.0, 10.2 Hz, 1H), 5.33–5.30 (m, 1H), 5.03 (dd, J = 10.2, 2.1 Hz, 1H), 4.92 (dd, J = 17.1, 2.1 Hz, 1H), 4.73 (dd, J = 8.9, 5.4 Hz, 1H), 4.33 (d, J = 6.7 Hz, 1H), 2.70 (ddd, J = 10.2, 6.7, 5.4 Hz, 1H), 1.52 (d, J = 1.1 Hz, 3H), 1.42 (d, J = 1.2 Hz, 3H), 1.19–1.11 (m, 21H); 13C NMR (151 MHz, C6D6) δ: 174.0, 138.9, 131.3, 120.4, 119.4, 75.2, 71.8, 51.7, 25.3, 17.9, 17.73, 17.68, 12.1; IR (film) ν: 3080, 2943, 2867, 1784, 1676, 1464, 1162, 882, 687 cm−1; HRMS (ESI-TOF) m/z calcd for C19H34O3SiNa [M + Na+] 361.2175. Found 361.2169.
:1; 2.8 mL).
3,5-Diisopropyl-4-vinyldihydrofuran-2(3H)-ones (17). NOTE: the title compounds are volatile. Therefore, in the cyclization step CH2Cl2 was used instead of DCE and the reaction mixture was heated in a sealed tube at 90 °C under argon for 1 h. The crude products were chromatographed on silica gel using Et2O/n-pentane mixture and the solvents were removed in a stream of argon.
(3R*,4R*,5R*)-3,5-Diisopropyl-4-vinyldihydrofuran-2(3H)-one (17a). Yield: 7.6 mg (33%); colorless crystals; mp: 64.3–65.5 °C; Rf (10% Et2O/n-pentane) 0.70; 1H NMR (600 MHz, CDCl3) δ: 5.82 (dt, J = 16.7, 10.1 Hz, 1H), 5.17–5.13 (m, 2H), 4.07 (dd, J = 8.4, 6.6 Hz, 1H), 3.00–2.95 (m, 1H), 2.27 (dd, J = 6.4, 4.3 Hz, 1H), 2.15–2.06 (m, 1H), 1.92–1.84 (m, 1H), 1.07–1.04 (m, 6H), 1.02 (d, J = 6.5 Hz, 3H), 0.90 (d, J = 6.7 Hz, 3H); 13C NMR (151 MHz, CDCl3) δ: 177.9, 135.3, 117.6, 86.4, 53.8, 45.9, 29.1, 28.5, 20.3, 20.1, 19.2, 18.3; IR (film) ν: 2960, 2925, 2874, 2853, 1771, 1467, 1200, 1176, 996, 723; HRMS (ESI-TOF) m/z calcd for C12H20O2Na [M + Na+] 219.1361. Found 219.1362.
(3S*,4R*,5R*)-3,5-Diisopropyl-4-vinyldihydrofuran-2(3H)-one (17b). Yield: 7.2 mg (31%); colorless oil; Rf (10% Et2O/n-pentane) 0.65; 1H NMR (600 MHz, CDCl3) δ: 5.60 (dt, J = 17.0, 10.6 Hz, 1H), 5.28 (dd, J = 10.3, 1.4 Hz, 1H), 5.23 (dd, J = 17.0, 1.4 Hz, 1H), 3.75 (dd, J = 10.6, 3.9 Hz, 1H), 3.04 (ddd, J = 10.5, 6.2, 3.9 Hz, 1H), 2.30 (dd, J = 10.3, 6.2 Hz, 1H), 1.86–1.73 (m, 2H), 1.23 (d, J = 6.5 Hz, 3H), 1.06 (d, J = 6.5 Hz, 3H), 0.91 (d, J = 6.6 Hz, 3H), 0.82 (d, J = 6.6 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ: 176.9, 131.1, 120.0, 86.4, 52.9, 47.9, 28.6, 25.6, 20.8, 20.6, 20.0, 16.7; IR (film) ν: 3508, 3088, 2959, 1760, 1643, 1474, 1364, 1173, 980, 773; HRMS (ESI-TOF) m/z calcd for C12H20O2Na [M + Na+] 219.1361. Found 219.1361.
(3R*,4R*,5S*)-3-Isopropyl-5-phenyl-4-vinyldihydrofuran-2(3H)-one (18a). Yield: 4.3 mg (16%); colorless oil; Rf (10% Et2O/hexane) 0.45; 1H NMR (600 MHz, C6D6) δ: 7.09–7.00 (m, 3H), 6.99–6.95 (m, 2H), 5.03 (d, J = 8.4 Hz, 1H), 4.80–4.73 (m, 1H), 4.68 (dd, J = 17.0, 1.9 Hz, 1H), 4.62 (dd, J = 9.8, 1.9 Hz, 1H), 2.90–2.82 (m, 1H), 2.24 (dd, J = 10.5, 4.0 Hz, 1H), 2.05 (sepd, J = 7.0, 4.0, 1H), 0.99 (d, J = 6.9 Hz, 3H), 0.88 (d, J = 7.0 Hz, 3H); 13C NMR (151 MHz, C6D6) δ: 176.0, 136.8, 136.5, 128.1, 127.9, 126.0, 117.2, 80.3, 48.3, 47.2, 27.2, 19.2, 19.0; IR (film) ν: 3066, 3033, 2962, 2933, 1779, 1643, 1455, 1156, 1018, 708 cm−1; HRMS (ESI-TOF) m/z calcd for C15H18O2Na [M + Na+] 253.1204. Found 253.1202.
(3S*,4R*,5S*)-3-Isopropyl-5-phenyl-4-vinyldihydrofuran-2(3H)-one (18b). Yield: 13.6 mg (50%); colorless oil; Rf (10% Et2O/hexane) 0.40; 1H NMR (600 MHz, CDCl3) δ: 7.35–7.30 (m, 2H), 7.27–7.20 (m, 3H), 5.47 (d, J = 4.6 Hz, 1H), 5.38 (dt, J = 16.9, 10.4 Hz, 1H), 4.95 (dd, J = 10.4, 1.2 Hz, 1H), 4.90 (dd, J = 16.9, 1.2 Hz, 1H), 3.32 (ddd, J = 10.9, 6.3, 4.6 Hz, 1H), 2.55 (dd, J = 10.2, 6.3 Hz, 1H), 1.94–1.85 (m, 1H), 1.28 (d, J = 6.5 Hz, 3H), 0.95 (d, J = 6.6 Hz, 3H); 13C NMR (151 MHz, CDCl3) δ: 176.5, 136.0, 131.0, 128.2, 127.7, 125.5, 120.3, 81.5, 52.7, 51.1, 25.8, 21.0, 20.6; IR (film) ν: 3068, 3033, 2960, 2872, 1775, 1644, 1454, 1160, 1125, 984, 698 cm−1; HRMS (ESI-TOF) m/z calcd for C15H18O2Na [M + Na+] 253.1204. Found 253.1203.
(3R*,4R*,5R*)-3-Isopropyl-5-phenyl-4-vinyldihydrofuran-2(3H)-one (18c). Yield: 3.8 mg (14%); colorless oil; Rf (10% Et2O/hexane) 0.30; 1H NMR (600 MHz, CDCl3) δ: 7.40–7.28 (m, 5H), 5.79 (ddd, J = 17.1, 10.2, 8.6 Hz, 1H), 5.17 (dd, J = 10.2, 0.5 Hz, 1H), 4.99 (m, 1H), 4.96 (d, J = 9.4 Hz, 1H), 2.89–2.82 (m, 1H), 2.63 (dd, J = 11.4, 3.6 Hz, 1H), 2.27 (sepd, J = 6.9, 3.6 Hz, 1H), 1.08 (d, J = 6.9 Hz, 3H), 1.04 (d, J = 7.0 Hz, 3H); 13C NMR (151 MHz, CDCl3) δ: 176.5, 137.8, 135.7, 128.5, 128.5, 125.9, 119.4, 82.8, 52.2, 52.0, 27.3, 19.5, 19.2; IR (film) ν: 3067, 3034, 2962, 2875, 1771, 1644, 1457, 1277, 1167, 977, 700 cm−1; HRMS (ESI-TOF) m/z calcd for C15H18O2Na [M + Na+] 253.1204. Found 253.1203.
TIPS removal. TIPS-protected lactone (0.01 mmol) was dissolved in anhydrous THF (0.5 mL) and the solution was cooled to 0 °C, before TBAF (1.0 M solution in THF; 0.012 mmol; 1.2 eq.) was added dropwise. The mixture was allowed to warm to 25 °C and after 10 min the reaction was stopped by the addition of saturated NH4Cl (0.5 mL). The aqueous phase was then extracted with Et2O (3 × 1.5 mL) and the combined organic layers were washed with brine (2 × 0.5 mL), dried over MgSO4 and evaporated. The residue was filtered through a pad of silica gel eluting with acetone/hexane and the solvent was removed. The deprotection product was directly used without further purification.
Formation of Mosher's ester. The deprotection product (0.01 mmol), triethylamine (5.6 μL, 0.04 mmol; 4 eq.) and 4-DMAP (0.3 mg; 0.0025 mmol; 0.25 eq.) were dissolved in anhydrous CH2Cl2 and (+)-MTPACl (1 M solution in CH2Cl2; 11 μL, 0.011 mmol; 1.1 eq.) was added dropwise at 25 °C. After stirring for 1 hour at the same temperature, the reaction was quenched with an aqueous NH4Cl (0.5 mL) solution. The resulting mixture was poured into water (10 mL) and the aqueous phase was extracted with CH2Cl2 (3 × 5 mL). The combined organic layers were washed with brine (15 mL), dried over MgSO4 and the solvent was removed in vacuo. Filtration of the crude product through a short plug of silica gel eluting with an acetone/hexane mixture afforded Mosher's ester.
(3S,4R,5S)-5-Isopropyl-2-oxo-4-vinyltetrahydrofuran-3-yl-(S)-3,3,3-trifluoro-2-methoxy-2-phenylpropanoate (19) and (3R,4S,5R)-5-isopropyl-2-oxo-4-vinyltetrahydrofuran-3-yl-(S)-3,3,3-trifluoro-2-methoxy-2-phenylpropanoate (19′). Obtained according to general procedure using 5a; inseparable mixture; yield: 3.0 mg (77%; 19/19′ = 1/1); colorless oil; Rf (10% acetone/hexane) 0.45; 19F NMR (376 MHz, CDCl3) δ: −71.68 (19′), −72.10 (19); HRMS (ESI-TOF) m/z calcd for C19H21F3O5Na [M + Na+] 409.1239. Found 409.1235.
(3S,4R,5S)-5-Isopropyl-2-oxo-4-vinyltetrahydrofuran-3-yl-(S)-3,3,3-trifluoro-2-methoxy-2-phenyl-propanoate (19). Obtained according to general procedure using (−)-5a. Yield 2.9 mg (74%); colorless oil; Rf (10% acetone/hexane) 0.45; ee > 99%; 19F NMR (376 MHz, CDCl3) δ: −71.68 (19′), −72.10 (major signal, 19); HRMS (ESI-TOF) m/z calcd for C19H21F3O5Na [M + Na+] 409.1239. Found 409.1235.
(3S,4S,5S)-2-Oxo-5-phenyl-4-vinyltetrahydrofuran-3-yl-(S)-3,3,3-trifluoro-2-methoxy-2-phenylpropanoate (20) and (3R,4R,5R)-2-oxo-5-phenyl-4-vinyltetrahydrofuran-3-yl-(S)-3,3,3-trifluoro-2-methoxy-2-phenylpropanoate (20′). Obtained according to general procedure using 10b; inseparable mixture: yield: 2.6 mg (61%; 20/20′ = 1/1); colorless oils; Rf (20% acetone/hexane) 0.50 and 0.45; 19F NMR (376 MHz, CDCl3) δ: −71.58 (20′), −72.11 (20); HRMS (ESI-TOF) m/z calcd for C19H21F3O5Na [M + Na+] 443.1082. Found 443.1074.
(3S,4S,5S)-2-Oxo-5-phenyl-4-vinyltetrahydrofuran-3-yl-(S)-3,3,3-trifluoro-2-methoxy-2-phenyl-propanoate (20). Obtained according to general procedure using (−)-10b. Yield: 3.5 mg (83%); colorless oil; Rf (20% acetone/hexane) 0.45; ee > 99%; 19F NMR (376 MHz, CDCl3) δ: −71.58 (20′), −72.11 (major signal, 20); HRMS (ESI-TOF) m/z calcd for C22H19F3O5Na [M + Na+] 443.1082. Found 443.1078.
Conclusions
In conclusion, we have demonstrated for the first time that the regioselectivity of allylation of aldehydes by ε-amidoallylindiums generated from β-lactams in the presence of InI and Pd(PPh3)4 can be reversed by the addition of a Brønsted acid. In the course of the process isomerisation of the initially formed linear α-adducts into their branched γ-isomers was observed, which is unprecedented reactivity of allylmetals. Obtained 1,3-diols and homoallylic alcohols can be easily converted without isolation into highly desirable γ-butyrolactones with three contiguous stereogenic centers, which are found in a broad range of natural products and their synthetic analogs exhibiting interesting pharmacological activities.3 Importantly, when enantioenriched substrates were used, expected γ-butyrolactones were obtained with high enantiomeric excess, demonstrating that the developed method may be applied in asymmetric synthesis, since a variety of β-lactams are readily available in both enantiomeric forms in excellent optical purity.1 Further elaboration of the chemistry presented and its application in the synthesis of selected natural products is currently in progress.Acknowledgements
Financial support of this research provided by Foundation for Polish Science, grant HOMING PLUS/2013-0/14 is gratefully acknowledged.Notes and references
- (a) R. J. Ternansky and J. M. Morin Jr, in The Organic Chemistry of β-Lactams, ed. G. I. Georg, VCH, New York, 1993, p. 257 Search PubMed; (b) A. Brandi, S. Cicchi and F. M. Cordero, Chem. Rev., 2008, 108, 3988 CrossRef CAS PubMed; (c) C. R. Pitts and T. Lectka, Chem. Rev., 2014, 114, 7930 CrossRef CAS PubMed; (d) A. Kamath and I. Ojima, Tetrahedron, 2012, 68, 10640 CrossRef CAS PubMed; (e) P. A. Magriotis, Eur. J. Org. Chem., 2014, 13, 2647 CrossRef; (f) N. Fu and T. T. Tidwell, Tetrahedron, 2008, 64, 10465 CrossRef CAS.
- (a) I. Ojima, in The Organic Chemistry of β-Lactams, ed. G. I. Georg, VCH, New York, 1993, p. 197 Search PubMed; (b) I. F. Ojima and F. Delaloge, Chem. Soc. Rev., 1997, 26, 377 RSC; (c) A. R. A. S. Deshmukh, B. M. Bhawal, D. Krishnaswamy, V. V. Govande, B. A. Shinkre and A. Jayanthi, Curr. Med. Chem., 2004, 11, 1889 CrossRef CAS PubMed; (d) B. Alcaide and P. Almendros, Curr. Med. Chem., 2004, 11, 1921 CrossRef CAS PubMed; (e) B. Alcaide, P. Almendros and C. Aragoncillo, Chem. Rev., 2007, 107, 4437 CrossRef CAS PubMed; (f) I. Ojima, E. S. Zuniga and J. D. Seitz, Top. Heterocycl. Chem., 2013, 30, 1 CAS.
- U. K. Klimczak and B. K. Zambroń, Chem. Commun., 2015, 51, 6796 RSC.
- (a) J. Peed, I. R. Davies, L. R. Peacock, J. E. Taylor, G. Kociok-Köhn and S. D. Bull, J. Org. Chem., 2012, 77, 543 CrossRef CAS PubMed , and refefences therein; (b) I. R. Davies, M. Cheeseman, R. Green, M. F. Mahon, A. Merritt and S. D. Bull, Org. Lett., 2009, 11, 2896 CrossRef CAS PubMed; (c) T. Classen, M. Korpak, M. Schölzel and J. Pietruszka, ACS Catal., 2014, 4, 1321 CrossRef CAS; (d) T. H. Al-Tel, R. A. Al-Qawasmeh, S. S. Sabri and W. Voelter, J. Org. Chem., 2009, 74, 4690 CrossRef CAS PubMed; (e) J. Priest, M. R. Longland, M. R. J. Elsegood and M. C. Kimber, J. Org. Chem., 2013, 78, 3476 CrossRef CAS PubMed.
- (a) H. Yorimitsu and K. Oshima, Bull. Chem. Soc. Jpn., 2009, 82, 778 CrossRef CAS; (b) H. Miyabe, Y. Yamaoka, T. Naito and Y. Takemoto, J. Org. Chem., 2003, 68, 6745 CrossRef CAS PubMed; (c) P. Jones and P. Knochel, J. Org. Chem., 1999, 64, 186 CrossRef CAS PubMed; (d) M. Sai, H. Yorimitsu and K. Oshima, Angew. Chem., Int. Ed., 2011, 50, 3294 CrossRef CAS PubMed; (e) S. Hayashi, K. Hirano, H. Yorimitsu and K. Oshima, Org. Lett., 2005, 7, 3577 CrossRef CAS PubMed; (f) S. Hayashi, K. Hirano, H. Yorimitsu and K. Oshima, J. Am. Chem. Soc., 2006, 128, 2210 CrossRef CAS PubMed; (g) Y. Takada, S. Hayashi, K. Hirano, H. Yorimitsu and K. Oshima, Org. Lett., 2006, 8, 2215 CrossRef PubMed; (h) L.-M. Zhao, H.-S. Jin, L.-J. Wan and L.-M. Zhang, J. Org. Chem., 2011, 76, 1831 CrossRef CAS PubMed.
- (a) J. Nokami, K. Yoshizane, H. Matsuura and S. Sumida, J. Am. Chem. Soc., 1998, 120, 6609 CrossRef CAS; (b) S. Sumida, M. Ohga, J. Mitani and J. Nokami, J. Am. Chem. Soc., 2000, 122, 1310 CrossRef CAS; (c) K.-T. Tan, S.-S. Chng, H.-S. Cheng and T.-P. Loh, J. Am. Chem. Soc., 2003, 125, 2959 Search PubMed; (d) T.-P. Loh, K.-T. Tan and Q.-Y. Hu, Angew. Chem., Int. Ed., 2001, 40, 2921 CrossRef CAS; (e) T.-P. Loh, Q.-Y. Hu and L.-T. Ma, Org. Lett., 2002, 4, 2389 CrossRef CAS PubMed; (f) H.-S. Cheng and T.-P. Loh, J. Am. Chem. Soc., 2003, 125, 4990 CrossRef CAS PubMed; (g) C.-L. K. Lee, C.-H. A. Lee, K.-T. Tan and T.-P. Loh, Org. Lett., 2004, 6, 1281 CrossRef CAS PubMed.
- L. Kuznetsova, I. M. Ungureanu, A. Pepe, I. Zanardi, X. Wu and I. Ojima, J. Fluorine Chem., 2004, 125, 487 CrossRef CAS.
- S. Araki and T. Hirashita, in Main Group Metals in Organic Synthesis, ed. H. Yamamoto and K. Oshima, WILEY-VCH Verlag GmbH & Co. KGaH, 2004, p. 323 Search PubMed.
- (a) Y. Takemoto, M. Anzai, R. Yanada, N. Fujii, H. Ohno and T. Ibuka, Tetrahedron Lett., 2001, 42, 1725 CrossRef CAS; (b) M. Anzai, R. Yanada, N. Fujii, H. Ohno, T. Ibuka and Y. Takemoto, Tetrahedron, 2002, 58, 5231 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Full characterization data and copies of NMR spectra for all new compounds. CCDC 1429878. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra00517a |
| This journal is © The Royal Society of Chemistry 2016 |