
Tumor-targeted docetaxel-loaded hyaluronic acid-quercetin polymeric micelles with p-gp inhibitory property for hepatic cancer therapy†
Abstract
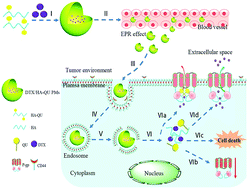
Docetaxel (DTX) has profound effects on several hepatic cancer (HC) cells, but shows unsatisfactory clinical efficacy due to the lack of tumor specificity and p-gp mediated drug efflux. Herein, a novel targeted drug delivery nanosystem based on hyaluronic acid (HA) and quercetin (QU) was designed to improve the in vivo therapeutic efficacy of DTX on HC through HA-CD44 mediated targeting and QU-based p-gp efflux inhibition. DTX-loaded HA–QU polymeric micelles (DTX/HA–QU PMs) displayed a mean particle size of 176.8 ± 3.4 nm with a low polydispersity index (<0.2). The drug loading and entrapment efficiency were 23.6 ± 1.3% and 86.8 ± 1.8%, respectively. Not only did the nanosystem facilitate cellular uptake via HA-CD44 mediated endocytosis, but it also preferentially accumulated at the tumor site via the EPR effect. Moreover, DTX/HA–QU PMs showed prolonged circulation time and high stability in the bloodstream, and achieved AUC0–∞ and t1/2 values that were 3.0-fold and 5.51-fold higher than those of Taxotere®, respectively. In addition, an in vitro cytotoxicity study showed that DTX/HA–QU PMs were 13.6-fold more effective than Taxotere®, judging by the IC50. Importantly, DTX/HA–QU PMs presented the highest antitumor efficacy in a xenograft tumor-bearing mice model and the tumor inhibition ratio was 73.91%. Meanwhile, DTX/HA–QU PMs could down-regulate p-gp expression in tumor cells. These results suggested that the targeting moiety and p-gp efflux inhibitory property help DTX to accumulate at the tumor site and improve its antitumor efficacy in vivo.
 Please wait while we load your content...
Please wait while we load your content...

