
High-yielding aqueous synthesis of chloroacetophenones and aroyl chlorohydrins†
Abstract
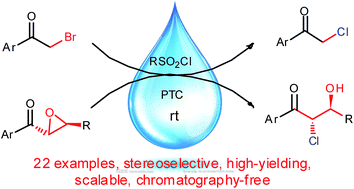
The use of large amounts of volatile organic solvents in industrial chemical processes contributes to widespread environmental pollution. To help solve this problem, water and a phase transfer catalyst were used to replace organic solvents in the transformations of bromoacetophenones into chloroacetophenones and aroyl epoxides into aroyl chlorohydrins. The reactions were promoted by sulfonyl chlorides and gave quantitative or close to quantitative yields. Notably, chromatographic purification, which is laborious and consumes large amounts of organic solvents, was not needed. These two processes have opened a green and cost-effective channel to prepare the chemical intermediates chloroacetophenones and aroyl chlorohydrins. The reaction mechanisms are discussed based on control experiments.
 Please wait while we load your content...
Please wait while we load your content...

