
Tailoring of spectral response and intramolecular charge transfer in β-enaminones through band gap tuning: synthesis, spectroscopy and quantum chemical studies†
Abstract
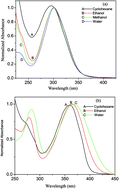
In this paper, we investigate the synthetic tailoring of the spectral response and intramolecular charge transfer (ICT) of β-enaminones through bandgap modulation. Two donor/acceptor substituted β-enaminones, namely, 3-(4-methoxyphenylamino)-2-cyclohexen-1-one (OACO) and 3-(4-nitrophenylamino)-2-cyclohexen-1-one (NACO) have been synthesized along with their unsubstituted counterpart, 3-(phenylamino)-2-cyclohexen-1-one (PACO). Steady state as well as time resolved spectroscopic techniques with picosecond resolution are used to record their spectral responses. Substitution of the donor group (–OCH3) mildly enhances the charge transfer from the phenyl ring to the enaminone moiety, while substitution of the acceptor group (–NO2) jeopardizes the charge transfer through mutual electron withdrawing effects of PNA and enaminone moieties. Combined experimental and quantum chemical investigations reveal that the ground state photophysics of OACO and NACO in water are controlled by both microscopic and macroscopic solvation with dominant contribution from the former. Time dependent density functional theory (TDDFT) calculations predict that the HOMO to [LUMO+1] transition gives rise to the absorption spectra of OACO in water, while the absorption by the enaminone moiety of NACO arises as a result of a HOMO to LUMO transition. A crossing between the first (S1) and the second excited (S2) states takes place in the microclusters of PACO, OACO and NACO with water. The intersystem crossing (ISC) has been found to be the major reason for low quantum yields in these molecules. The band gap modulation through waxing and waning of the conjugation strength is expected to throw light on many ICT-driven processes and provides means of tuning the properties depending on it.
 Please wait while we load your content...
Please wait while we load your content...

