
Isolation, resolution and biological evaluation of pestalachlorides E and F containing both point and axial chirality†
Qian Xing‡
ac,
Li-She Gan‡b,
Xiao-Feng Mou‡a,
Wei Wanga,
Chang-Yun Wangad,
Mei-Yan Weia and
Chang-Lun Shao*ad
aKey Laboratory of Marine Drugs, The Ministry of Education of China, School of Medicine and Pharmacy, Ocean University of China, Qingdao 266003, People's Republic of China. E-mail: shaochanglun@163.com
bCollege of Pharmaceutical Sciences, Zhejiang University, Hangzhou 310058, People's Republic of China
cHuantai Food and Drug Administration, Zibo 256400, People's Republic of China
dLaboratory for Marine Drugs and Bioproducts, Qingdao National Laboratory for Marine Science and Technology, Qingdao 266200, People's Republic of China
First published on 22nd February 2016
Abstract
Marine life forms are an important source of structurally diverse and biologically active natural products. As a unique case of both enantiomeric and atropisomeric isomers being present in one marine natural product structure, two new dichlorinated diphenylmethanes containing both point and axial chirality, (±)-pestalachlorides E (1) and F (2), were isolated from a marine-derived Pestalotiopsis (ZJ-2009-7-6) fungus. Both of them showed potent antifouling activities against the larval settlement of the barnacle Balanus amphitrite at nontoxic concentrations with EC50 values of 1.65 and 0.55 μg mL−1, respectively, and antifouling activity was detected for the first time for this class of metabolites.
Introduction
Over the past 50 years, more than 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 700 new marine natural compounds have been obtained from marine organisms collected from 1300 genera across 28 phyla.1,2 Marine natural products have proven to be rich sources of structurally novel and biologically active compounds that have become significant chemical entities for drug discovery.3,4 To date, there are seven marine natural products and thirteen marine natural products inspired compounds that are FDA-approved agents or in clinical trial.5
700 new marine natural compounds have been obtained from marine organisms collected from 1300 genera across 28 phyla.1,2 Marine natural products have proven to be rich sources of structurally novel and biologically active compounds that have become significant chemical entities for drug discovery.3,4 To date, there are seven marine natural products and thirteen marine natural products inspired compounds that are FDA-approved agents or in clinical trial.5
Marine-derived microorganisms have proved to be a rich and important source of drug candidates.1,6 Dichlorinated benzophenones (diphenylmethanes) represent a rare family of marine natural products that only six in the class have been reported to date, including pestalone,7 desmethyl pestalone,8 and pestalachlorides A–D.9,10 As part of a continuing program to evaluate bioactive marine natural products as potential drug leads from marine invertebrate derived microorganisms,11–14 further chemical investigation15,16 of laboratory cultures of the Pestalotiopsis fungus (ZJ-2009-7-6) resulted in the isolation of two new chlorinated enantiomeric diphenylmethanes, (±)-pestalachlorides E (1) and F (2) (Fig. 1). They both exist as a pair of separable enantiomers, which in turn display additional atropisomerism. The planar structures and relative configurations were first identified by comprehensive analysis of spectroscopic data, including 2D NMR and single crystal X-ray diffraction data. After chiral HPLC separation of (+)-1/(−)-1 and (+)-2/(−)-2, the inseparable atropisomeric features of (+)-1 and (−)-1, (+)-2 and (−)-2 caused by restricted rotation of the C8–C9 bond, were studied theoretically as M and P isomers by potential energy surface scans and energy barrier calculations at the B3LYP/6-31G level. Furthermore, DFT and TDDFT calculations of their CD and NMR spectra allowed the assignments of their absolute configurations. The isolation, structure elucidation, chiral resolution, and theoretical studies of the atropisomeric features of compounds 1 and 2 (Fig. 1) are presented herein, along with their potent antifouling activities against the larval settlement of barnacle Balanus amphitrite.
 | ||
| Fig. 1 Structures of 1 and 2 including key 1H–1H COSY, HMBC, and NOESY correlations. | ||
Results and discussion
Compound 1 (ref. 17) was obtained as a colourless crystal in acetone. Its molecular formula, C21H22O6Cl2 with 10 degrees of unsaturation, was determined by the HRESIMS peak at m/z 441.0857 (calcd C21H23O6Cl2, 441.0866). The IR absorption bands at 3398 and 1630 cm−1 suggested the presence of a hydroxy and a conjugated carbonyl groups, respectively. The 1H NMR spectrum showed duplicated signals in a ratio of about 6:1. Close examination of the NMR data allowed the interpretation and assignments of all protons to both isomers. For the major isomer, the existence of one hydrogen-bonded phenolic proton (δH 11.86, 1H, s), one aldehyde group (δH 9.58, 1H, s), one aromatic proton (δH 6.25, 1H, s), one methoxy group (δH 3.15, 3H, s), one aromatic methyl group (δH 2.42, 3H, s), two methine protons (δH 5.27 and 2.65), two methylene protons (δH 3.21 and 2.87), and two methyl groups (δH 1.26 and 1.20) were observed. The 13C NMR and DEPT spectra showed 21 carbon signals, including one aldehyde group (δC 193.8) and twelve aromatic carbons representing two phenyl rings, which took up nine of the ten degrees of unsaturation. The structure of 1 was then suggested to contain a third ring. Moreover, the position of the hydroxyisopropyl group was located at C-2′ in ring B supported by the HMBC correlations from H-1′ and H-2′ to C-3′, and from H-4′ and H-5′ to C-2′. Finally, these HMBC correlations from H-8 to C-9, C-10, and C-14, and from H-2′ to C-9 enabled the establishment of the C-8–C-9 linkage.
The structural architecture of 1 was then established by comprehensive analysis of 2D NMR spectroscopic data, especially 1H–1H COSY and HMBC correlations. The aromatic ring A and the five-membered ring B were established by 1H–1H COSY correlations of H-8/H-2′ and H-2′/H-1′ together with HMBC correlations from H-8 to C-2, C-6, and C-7, from H-1′ to C-5, C-6, and C-7, and from H-2′ to C-6 and C-7 (Fig. 1). According to the above data, the planar structure was established as shown in 1. Furthermore, the relative configuration of 1 was assigned as trans on the basis of the NOESY correlations between H-8 and the H3-4′ and H3-5′ of the two methyls on the side chain. For the minor component of 1, the same structure can be concluded. The most distinguishable differences in 1H NMR of the two isomers are the chemical shifts of the two methoxy groups at C-14 (CH3-16), indicating the possible atropisomerism in 1. It should be mentioned that the 13C NMR data of the minor component of 1 were not observed because of the minor quantity (Table 2).
| Position | 1 | 2 | ||
|---|---|---|---|---|
| Major | Minor | Major | Minor | |
| 1 | 9.58 (1H, s) | 9.73 (1H, s) | 9.49 (1H, s) | 9.60 (1H, s) |
| 4 | 6.25 (1H, s) | 6.17 (1H, s) | 6.21 (1H, s) | 6.28 (1H, s) |
| 8 | 5.27 (1H, d, 5.5) | 5.45 (1H, d, 5.5) | 5.11 (1H, d, 6.0) | 5.33 (1H, brs) |
| 15 | 2.42 (3H, s) | 2.42 (3H, s) | 2.43 (3H, s) | 2.43 (3H, s) |
| 16 | 3.15 (3H, s) | 4.13 (3H, s) | 3.89 (3H, s) | 3.17 (3H, s) |
| 1′ | 3.21 (1H, dd, 16.3, 10.1) | 3.29 (1H, overlapped) | 2.82 (1H, dd, 17.2, 7.5) | 2.82 (1H, dd, 17.2, 7.5) |
| 2.87 (1H, dd, 16.3, 4.2) | 2.87 (1H, overlapped) | 3.29 (1H, overlapped) | 3.29 (1H, overlapped) | |
| 2′ | 2.65 (1H, ddd, 10.1, 5.5, 4.2) | 2.65 (1H, overlapped) | 3.47 (1H, overlapped) | 3.27 (1H, overlapped) |
| 4′ | 1.26 (3H, s) | 1.26 (3H, s) | 4.72 (2H, s) | 4.72 (2H, s) |
| 5′ | 1.20 (3H, s) | 1.20 (3H, s) | 1.81 (3H, s) | 1.78 (3H, s) |
| 3-OH | 11.86 (H, brs) | 12.04 (H, brs) | 11.92 (1H, s) | 11.90 (1H, s) |
| 5-OH | — | — | 9.74 (1H, s) | 9.89 (1H, s) |
| 10-OH | — | — | 8.13 (1H, s) | 8.66 (1H, s) |
| Position | 1 | 2 | |
|---|---|---|---|
| Major | Major | Minor | |
| 1 | 193.8, CH | 192.9, CH | 193.6, CH |
| 2 | 111.8, C | 112.0, C | 112.1, C |
| 3 | 166.2, C | 165.8, C | 166.1, C |
| 4 | 101.7, CH | 101.4, CH | 101.6, CH |
| 5 | 162.7, C | 161.9, C | 162.3, C |
| 6 | 125.1, C | 123.9, C | 124.1, C |
| 7 | 152.9, C | 151.9, C | 152.3, C |
| 8 | 44.4, CH | 47.3, CH | 46.8, CH |
| 9 | 129.9, C | 126.7, C | 128.1, C |
| 10 | 149.2, C | 151.0, C | 151.2, C |
| 11 | 119.1, C | 119.0, C | 118.0, C |
| 12 | 134.7, C | 134.7, C | 134.8, C |
| 13 | 121.4, C | 120.5, C | 121.4, C |
| 14 | 155.4, C | 153.1, C | 155.5, C |
| 15 | 18.2, CH3 | 18.2, CH3 | 19.7, CH3 |
| 16 | 60.5, CH3 | 62.3, CH3 | 60.4, CH3 |
| 1′ | 32.2, CH2 | 34.6, CH2 | 34.7, CH2 |
| 2′ | 59.0, CH | 54.1, CH | 54.9, CH |
| 3′ | 73.6, C | 148.0, C | 148.4, C |
| 4′ | 28.2, CH3 | 112.3, CH2 | 110.4, CH2 |
| 5′ | 26.0, CH3 | 18.2, CH3 | 18.2, CH3 |
Compound 2 (ref. 18) was also obtained as a colorless crystal in acetone. Its HRESIMS data showed a molecular formula of C21H20Cl2O5 and one more degree of unsaturation than that in 1. The 1H and 13C NMR spectra of 2 also displayed duplicated signals in a ratio of approximately 5:4. Compared to the 1H NMR spectrum of 1, a singlet for a terminal double bond at δH 4.72 (2H, s) was exhibited replacing that methyl group in 1, which was also confirmed by two olefinic carbon signals resonating at δC 148.4 and 110.4 in its 13C NMR spectrum. Thus the gross structure of 2 was concluded as the 3′,4′-dehydration product of pestalachloride E (1). The presence of the C-3′–C-4′ double bond was further supported by the HMBC correlations from H-5′ to C-3′ and C-4′ (Fig. 1). Furthermore, the relative configuration between H-8 and H-2′ in 2 was also determined as trans on the basis of the NOESY data. Therefore, the structure of 2 was established and named as pestalachloride F. Similar to 1, the major and minor components in 2 may also be atropisomers.
Fortunately, by slow crystallization in acetone, single crystals of 1 suitable for X-ray diffraction analysis were obtained. The structure and the trans relative configuration of 1 was then confirmed by the single-crystal X-ray diffraction data19 (Fig. 2a). Further analysis of the X-ray data of 1 revealed that it possesses a centrosymmetric space group C2/c, indicating a racemic nature.
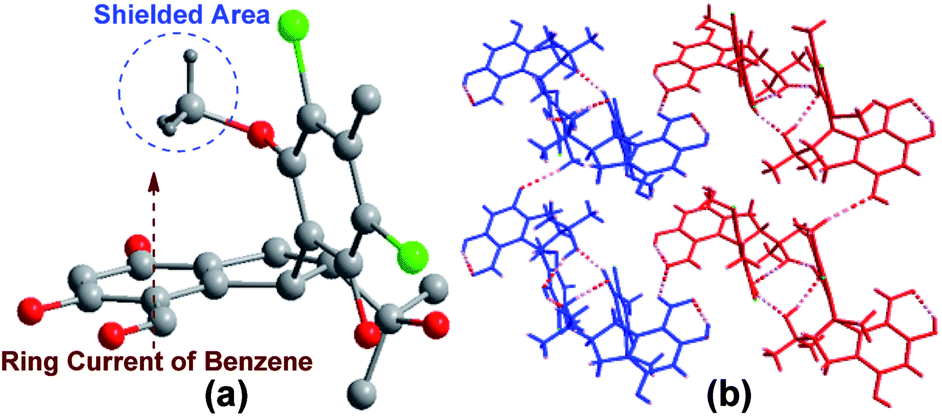 | ||
| Fig. 2 (a) Perspective ORTEP drawing for 1. (b) View of 2-D sheet structure. The broken lines represent the hydrogen bonds [O–H⋯O and C–H⋯O (blue)]. | ||
In order to fully assign the NMR data and clarify the racemic atropisomer nature of 1 and 2, HPLC analysis of 1 and 2 on a chiral column (Kromasil 5-TBB) were carried out firstly. Two distinct chromatographic peaks with a ratio of 1:1 were found and isolated from both 1 and 2. Subsequently, chemical shifts of the most distinguishable signal of H3-16 can be rationally explained by the shielding and deshielding zones of the benzene ring A (Fig. 2a) based on the X-ray structure. As shown in Fig. 2a for the M isomer X-ray 3D structure of 1, the methoxy group situates directly over the shielded area of aromatic ring A. Meanwhile, intramolecular hydrogen bonds between OH-10 and OH-3′, which can be observed in X-ray data (Fig. 2b), may also stabilize this M isomer. Thirdly, variable temperature NMR experiments has shown that these minor atropisomers can be transformed to the corresponding major one at 75 °C in DMSO-d6 (Fig. S13†). Because the M isomer in (−)-1 and the P isomer in (+)-1 are enantiomers, full assignments still cannot be done without the assignment of absolute configuration.
The absolute configurations of (+)-1, (−)-1, (+)-2, and (−)-2 as well as the inseparable P and M atrop-diastereomers were studied theoretically. Conformational analysis was carried out for 1 and 2 via Monte Carlo searching using molecular mechanism with MMFF94 force field in the SPARTAN 08 software package. A 1D PES scan on the dihedral angle of 10-9-8-2′ in representing conformer of M isomers of 1 and 2 were then performed by modredundant optimization at the semi-empirical AM1 level in Gaussian 09 software package.20 The resulted transitional state conformers and all the lowest energy conformers were re-optimized using DFT at the B3LYP/6-31+G(d) level in gas phase. All the ΔG (relative Gibbs energy barrier) for M–P conversion is greater than 20 kcal mol−1, which confirmed the existence of M/P isomers at room temperature (Fig. 4).21 Subsequently, TD DFT calculations of their ECD spectra showed that the inseparable M and P atrop-diastereomers have similar theoretical ECD spectra, which gave the possibility for the determination of the absolute configurations at C-8 and C-2′ of the chiral column isolates. As shown in Fig. 3, the calculated 8S, 2′R configuration of compounds 1 and 2 showed first positive and second negative Cotton effects, and correspondingly, the experimental ECD spectra of (−)-1 and (−)-2 also exhibited curves with the same pattern, which indicated the absolute configuration of (−)-1 and (−)-2 as 8S,2′R. The 8R,2′S configuration was then assigned to compounds (+)-1 and (+)-2.
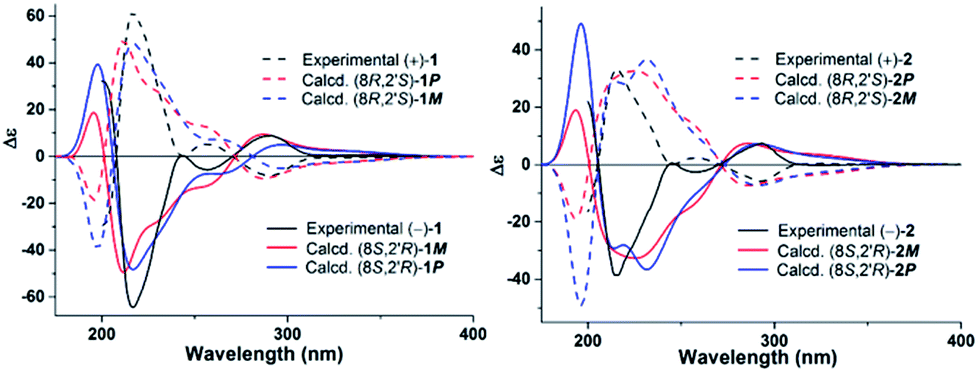 | ||
| Fig. 3 Experimental ECD spectra (black lines) and calculated ECD spectra of M and P atropisomers (red and blue lines) of 1 and 2. | ||
 | ||
| Fig. 4 Atropisomeric structures of 1 and 2. | ||
As indicated by the 1H NMR integration values of the M and P atrop-diastereomers, their ratio are 6:1 in (−)-1 and (+)-1, and 5:4 in (−)-2 and (+)-2 (Table 1). The major and minor components in the atrop-diastereomers were assigned based on their relative Gibbs free energy and Boltzmann distribution theory, as well as NMR theoretical studies of the four representative lowest energy conformers. To predict the ratio of M and P diastereomers in (−)-1 and (−)-2, all the calculated lowest energy M and P conformers were weighted together in the Boltzmann function by their relative Gibbs free energy and calculated separately in two groups of M and P, the results showed a ratio of P:M = 2.73:1 for (−)-1, and P:M = 1:2.64 for (−)-2. Based on the above conclusion, the major component in (−)-1 is the P isomer, while the M isomer is the major component in (−)-2. The assignments were further confirmed by NMR calculation. As indicated in Table S1,† the theoretical chemical shifts at CH3-16 of four representative lowest energy conformers, (8S,2′R)-1M-C3, (8S,2′R)-1P-C8, (8S,2′R)-2M-C1, and (8S,2′R)-2P-C1 were again in good agreements with the experimental data (Fig. S28–31†). Because the M isomer in (−)-1 and P isomer in (+)-1 are enantiomers, and (+)-1 and (−)-1 show the same 1H NMR spectra, the ratio of P and M atropisomers in (+)-1 should be just on the contrary of (−)-1. The same conclusion also can be drawn for (+)-2. The ratio of P and M atropisomers in 2 is obviously different from that in 1, which may be caused by the loss of intramolecular hydrogen bond.
Compounds (±)-1 and (±)-2 were also evaluated for antifouling activity against the larval settlement of the barnacle Balanus amphitrite.22 Both of them showed potent antifouling activities at nontoxic concentrations with EC50 values of 1.65 and 0.55 μg mL−1 (SeaNine 211 as a positive control, IC50 = 1.23 μg mL−1, LC50/EC50 = 20.3), respectively, which were much lower than the standard requirement of an EC50 of 25 μg mL−1 established by the U.S. Navy program as an efficacious level for natural antifouling agents. To the best of our knowledge, this is the first report of antifouling activities for this class of metabolites. Furthermore, a compound with therapeutic ratio (LC50/EC50) > 15 is often considered to be a nontoxic antifouling compound.23 Compounds (±)-1 and (±)-2 have high therapeutic ratios LC50/EC50 > 30.3 and 18.2, respectively, which were higher than 15, suggesting that they might be regarded as environmentally benign antifouling agents.
Experimental section
General experimental procedures
Melting points were determined on an X-6 micromelting point apparatus and are uncorrected. Optical rotations were measured on a JASCO P-1020 digital polarimeter. UV spectra were obtained on a Beckman DU 640 spectrophotometer. IR spectra were recorded on a Bruker EQUINOX 55 spectrometer using KBr pellets. NMR spectra were recorded on an Agilent DD2 NMR spectrometer (500 MHz for 1H and 125 MHz for 13C). Chemical shifts δ are reported in ppm, using TMS as internal standard and coupling constants (J) are in Hz. Single-crystal data was measured on an Agilent Gemini ultra diffractometer (Cu Kα radiation). Electrospray ionization mass spectrometry (ESIMS) and high resolution electrospray ionization mass spectrometry (HRESIMS) spectra were obtained on a Micromass Q-TOF spectrometer. HPLC analysis and separation was performed in a Waters 1525 preparative HPLC system coupled with Waters 2996 photodiode array detector. A Kromasil 100-5-C18 preparative HPLC column (250 × 10 mm, 82599) and a Kromasil 5-TBB preparative HPLC column (250 × 10 mm, E65007) were used. Silica gel (Qing Dao Hai Yang Chemical Group Co.; 200–300 mesh) and Sephadex LH-20 (GE Healthcare) were used for column chromatography (CC). Precoated silica gel plates (Yan Tai Jiang You Silica Gel Development Limited Company) were used for thin layer chromatography (TLC).Fungal material
The fungal strain Pestalotiopsis sp. (ZJ-2009-7-6) was isolated from a piece of fresh tissue from the inner part of a soft coral Sarcophyton sp., collected from Yongxing Island in the South China Sea in November, 2009. The strain was identified as Pestalotiopsis sp. according to morphologic traits and molecular identification. Its 617 base pair ITS sequence had 99% sequence identity to that of Pestalotiopsis sp. DFFW (EF055190). The sequence data have been submitted to GenBank with the accession number HM486429.Fermentation and extraction
The fungal strain Pestalotiopsis sp. (ZJ-2009-7-6) was cultivated on solid rice medium (composition of artificial seawater: sea salt 33 g in 1 L distilled water; composition of medium: parboiled rice in bromine-modified artificial seawater, in 1 L Erlenmeyer flask each containing 30 g rice and 45 mL artificial seawater.) in 60 Erlenmeyer flasks at 27 °C in static for 4 weeks. Then the culture was extracted three times with an equal volume of CHCl3/MeOH (v/v, 1:1). The organic extracts were combined and concentrated under vacuum to afford a dry crude extract.
Isolation
The resulting exact (5.4 g) was subjected to silica gel column chromatography (CC) (petroleum ether, EtOAc v/v, gradient elution) and offered 5 fractions (Fr.1–Fr.5). Fr.2 was subjected to silica gel column chromatography (CC) (petroleum ether, EtOAc v/v, gradient elution) and yielded 5 fractions (Fr.21–Fr.25). Fr.25 was further purified by semi-preparative HPLC eluting with 85% of MeOH/H2O at a flow rate of 2.0 mL min−1 to give 2 (1.9 mg). Fr.4 was subjected to silica gel column chromatography (CC) (petroleum ether, EtOAc v/v, gradient elution) and produced 6 fractions (Fr.41–Fr.46). Fr.44 was subjected to Sephadex LH-20 CC eluting with mixtures of CHCl3–MeOH (v/v, 1:1) and purified repeatedly by semi-preparative HPLC eluted with 68% of MeOH/H2O at a flow rate of 2.0 mL min−1 to give 1 (3.2 mg). Compound 1 was then obtained by chiral preparative HPLC eluting with 85% of n-hexane/isopropanol at a flow rate of 2.0 mL min−1 to give (+)-1 (1.7 mg) and (−)-1 (1.5 mg). Similarly, 2 was separated by chiral preparative HPLC eluting with 95% of n-hexane/isopropanol at a flow rate of 2.0 mL min−1 to give (+)-2 (0.9 mg) and (−)-2 (1.0 mg).
X-ray crystallographic analysis of 1
Colorless crystals of 1 were obtained from acetone. The crystal data was recorded at 293 K on a Bruker APEX-II CCD diffractometer with Cu Kα radiation (λ = 1.54178 Å). The structure was solved by direct methods (SHELXS-97) and refined using full-matrix least-squares difference Fourier techniques. The crystallographic data of 1 have been deposited at the Cambridge Crystallographic Data Centre with the deposition number 1052426.ECD and NMR spectra calculation
In general, conformational analyses were carried out via Monte Carlo searching using molecular mechanism with MMFF94 force field in the SPARTAN 08 software package. The resulted lowest energy conformers were re-optimized using DFT at the B3LYP/6-31+G(d) level in gas phase by using the GAUSSIAN 09 program. The B3LYP/6-31+G(d) harmonic vibrational frequencies were also calculated to confirm their stability. The energies, oscillator strengths, and rotational strengths (velocity) of the first 60 electronic excitations of each lowest energy conformers were calculated using the TDDFT methodology at the B3LYP/6-311++G(2d,2p) level in vacuum. The ECD spectra were then simulated by the overlapping Gaussian function (half the bandwidth at 1/e peak height, σ = 0.3 eV). To get the overall spectra, the simulated spectra of the lowest energy conformers for each of the M and P atrop-isomers were averaged according to the Boltzmann distribution theory and their relative Gibbs free energy (ΔG). Theoretical ECD spectra of (8R,2′S)-1P, (8R,2′S)-1M, (8R,2′S)-2P, and (8R,2′S)-2M, were obtained by directly inverse of the ECD spectra of the corresponding enantiomers, (8S,2′R)-1M, (8S,2′R)-1P, (8S,2′R)-2M, and (8S,2′R)-2P, respectively. For NMR calculation, Gauge-Independent Atomic Orbital (GIAO) calculations of 1H and 13C NMR chemical shifts were accomplished by ab initio density functional theory (DFT) at the rmpw1pw91-SCRF (acetone)/6-311++g(2d,2p) level with the PCM solvent continuum model in Gaussian 09 software. The 1H and 13C NMR chemical shifts of TMS were calculated in the same level and used as the references.Antifouling activity
The antifouling activities against the larval settlement of the barnacle were determined using cyprids of Balanus amphitrite Darwin according to literature procedures.22Conclusions
Two new chlorinated enantiomeric diphenylmethanes containing both point and axial chirality, (±)-pestalachlorides E (1) and F (2), were studied experimentally and theoretically. After HPLC chiral separation of (+)-1/(−)-1 and (+)-2/(−)-2, the inseparable atropisomeric features of (+)-1 and (−)-1, (+)-2 and (−)-2 caused by restricted rotation of the C8–C9 bond, were studied theoretically and identified as M and P isomers. Moreover, (±)-1 and (±)-2 showed potent antifouling activities against the larval settlement of the barnacle B. amphitrite with EC50 values of 1.65 and 0.55 μg mL−1, respectively.Acknowledgements
We thank Prof. Yu-Cheng Gu (Syngenta) for his proofreading of the manuscript and Prof. Pei-Yuan Qian (HKUST) for antifouling bioassay. This work was supported by the Program of National Natural Science Foundation of China (No. 41322037 and 41130858), the Program of Natural Science Foundation of Shandong Province of China (No. JQ201510), the Marine Special Public Welfare Scientific Research, State Oceanic Administration of China (No. 201405038), and the Taishan Scholars Program, China.Notes and references
- J. W. Blunt, B. R. Copp, R. A. Keyzers, M. H. G. Munro and M. R. Prinsep, Nat. Prod. Rep., 2015, 32, 116–211 RSC.
- J. W. Blunt, B. R. Copp, R. A. Keyzers, M. H. G. Munro and M. R. Prinsep, Nat. Prod. Rep., 2014, 31, 160–258 RSC.
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2012, 75, 311–335 CrossRef CAS PubMed.
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2007, 70, 461–477 CrossRef CAS PubMed.
- W. H. Gerwick and B. S. Moore, Chem. Biol., 2011, 19, 85–98 CrossRef PubMed.
- X. M. Hou, R. F. Xu, Y. C. Gu, C. Y. Wang and C. L. Shao, Curr. Med. Chem., 2015, 22, 3707–3762 CrossRef CAS PubMed.
- M. Cueto, P. R. Jensen, C. Kauffman, W. Fenical, E. Lobkovsky and J. Clardy, J. Nat. Prod., 2001, 64, 1444–1446 CrossRef CAS.
- E. W. Li, L. H. Jiang, L. D. Guo, H. Zhang and Y. S. Che, Bioorg. Med. Chem., 2008, 16, 7894–7899 CrossRef CAS PubMed.
- N. Slavov, J. Cvengros, J. M. Neudorfl and H. G. Schmalz, Angew. Chem., Int. Ed., 2010, 49, 7588–7591 CrossRef CAS PubMed.
- M. Y. Wei, D. Li, C. L. Shao, D. S. Deng and C. Y. Wang, Mar. Drugs, 2013, 11, 1050–1060 CrossRef CAS PubMed.
- M. Chen, C. L. Shao, X. M. Fu, C. J. Kong, Z. G. She and C. Y. Wang, J. Nat. Prod., 2014, 77, 1601–1606 CrossRef CAS PubMed.
- C. L. Shao, R. F. Xu, M. Y. Wei, Z. G. She and C. Y. Wang, J. Nat. Prod., 2013, 76, 779–782 CrossRef CAS PubMed.
- K. L. Yang, M. Y. Wei, C. L. Shao, X. M. Fu, Z. Y. Guo, R. F. Xu, C. J. Zheng, Z. G. She, Y. C. Lin and C. Y. Wang, J. Nat. Prod., 2012, 75, 935–941 CrossRef CAS PubMed.
- C. L. Shao, H. X. Wu, C. Y. Wang, Q. A. Liu, Y. Xu, M. Y. Wei, P. Y. Qian, Y. C. Gu, C. J. Zheng, Z. G. She and Y. C. Lin, J. Nat. Prod., 2011, 74, 629–633 CrossRef CAS PubMed.
- Y. L. Jia, M. Y. Wei, H. Y. Chen, F. F. Guan, C. Y. Wang and C. L. Shao, Org. Lett., 2015, 17, 4216–4219 CrossRef CAS PubMed.
- Q. Xing, D. Li, Z. Y. Guo, C. Y. Wang and C. L. Shao, Chem. Nat. Compd., 2015, 51, 1080–1084 CrossRef CAS.
- Pestalachloride E (1): colorless crystals; mp 272–274 °C; UV (MeOH) λmax (logε) 209 (4.13), 292 (3.86), 341 (3.19) nm; IR (KBr) νmax 3398, 2925, 2855, 2369, 1630, 1381, 1282, 1140 cm−1; 1H NMR and 13C NMR, see Tables 1 and 2; ESI-MS m/z 441.2 [M + H]+, 423.1 [M + H − H2O]+; HRESI-MS m/z 441.0857 [M + H]+ (calcd for C21H23O6Cl2, 441.0866). (+)-Pestalachloride E [(+)-(1)]: [α]26D +24.0 (c 0.065, MeOH). (−)-Pestalachloride E [(−)-(1)]: [α]26D −20.0 (c 0.065, MeOH).
- Pestalachloride F (2): colorless platelet; mp 255–257 °C; UV (MeOH) λmax (logε) 210 (3.81), 292 (3.52), 337 (3.03) nm; IR (KBr) νmax 3484, 2364, 2342, 1631, 1377, 1289, 728 cm−1; 1H NMR and 13C NMR, see Tables 1 and 2; ESI-MS m/z 423.14 [M + H]+; HRESI-MS m/z 423.0759 [M + H]+ (calcd for C21H21O5Cl2, 423.0761). (+)-Pestalachloride F [(+)-(2)]: [α]26D +18.0 (c 0.045, MeOH). (−)-Pestalachloride F [(−)- (2)]: [α]26D −15.3 (c 0.045, MeOH).
- Crystal data for 1: C24H28O7Cl2, Mr = 499.36, monoclinic, space group C2/c with a = 19.1983(8) Å, b = 10.9513(4) Å, c = 22.9245(9) Å, β = 96.2213(0)°, V = 4791.4(3) Å3, Z = 8, Dx = 1.385 mg m−3, μ(Cu Kα) = 2.802 mm−1, and F(000) = 2096. Crystal dimensions: 0.66 × 0.34 × 0.26 mm3. Independent reflections: 3250 (Rint = 0.2062). The final R1 values were 0.0636, wR2 = 0.1662 (I > 2σ(I)). Crystallographic data have been deposited in the Cambridge Crystallographic Data Centre (deposition number CCDC 1052426).
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Rev. C 01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- S. R. LaPlante, P. J. Edwards, L. D. Fader, A. Jakalian and O. Hucke, ChemMedChem, 2011, 6, 505–513 CrossRef CAS PubMed.
- V. Thiyagarajan, T. Harder, J. W. Qiu and P. Y. Qian, Mar. Biol., 2003, 143, 543–554 CrossRef.
- N. Fusetani, Nat. Prod. Rep., 2011, 28, 400–410 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1052426. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra00374e |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2016 |