
Control of osmotic pressure through CO2-capture and release facilitated by the lower critical solution temperature (LCST) phase transition of acylated branched polyethylenimine†
Abstract
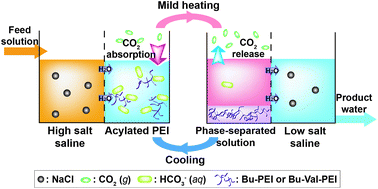
A solution of acylated polyethylenimine can absorb CO2 at low temperatures to elevate the osmotic pressure and draw water from a high-salt saline solution. Such a solution can be phase-separated to liberate CO2 by mild heating at 40 °C, and the osmotic pressure can be effectively reduced for water release into a low-salt saline solution. The osmotic pressure-controlling system has the potential to be used as a draw solution for forward osmosis.
 Please wait while we load your content...
Please wait while we load your content...

