
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Disturbed cofactor binding by a novel mutation in UDP-galactose 4′-epimerase results in a type III galactosemia phenotype at birth†
Stephanie
Paul
ab,
Thomas J.
McCorvie‡
a,
Johannes
Zschocke
c and
David J.
Timson
*ad
aSchool of Biological Sciences, Queen's University Belfast, Medical Biology Centre, 97 Lisburn Road, Belfast, BT9 7BL, UK
bInstitute for Global Food Security, Queen's University Belfast, 18-30 Malone Road, Belfast, BT9 5BN, UK
cDivision of Human Genetics, Innsbruck Medical University, Innsbruck 6020, Austria
dSchool of Pharmacy and Biomolecular Sciences, University of Brighton, Huxley Building, Lewes Road, Brighton BN2 4GJ, UK. E-mail: d.timson@brighton.ac.uk; Fax: +44(0) 1273 642090; Tel: +44(0) 1273 641623
First published on 3rd February 2016
Abstract
UDP-galactose 4′-epimerase (GALE) is an essential enzyme in galactose metabolism and its dysfunction results in type III galactosemia. Herein we report a patient born with abnormal blood galactose levels and reduced GALE activity who was shown to be heterozygous for a c.266C>T missense mutation in the GALE gene, predicted to result in the amino acid exchange p.A89V. Over a period of months, the patient's blood galactose, galactose 1-phosphate and GALE activity levels reverted to normal, encouraging us to investigate this mutation. Structurally Ala89 is a highly conserved residue located close to the binding site of the cofactor, NAD+. Consequentially molecular modelling predicted that this mutation results in steric clashes between the cofactor and valine side chain, and bioinformatic predictions suggested that p.A89V is likely to be less stable than the wild-type. Biochemical studies on the recombinant p.A89V enzyme demonstrated lower activity than the wild type (Km increased by approximately 30-fold; kcat reduced approximately 180-fold), and additionally changes in stability and altered NAD+ binding were observed. Thus, a picture emerges in which this mutation leads to reduced stability, disturbed cofactor binding and subsequently reduced activity. Overall this study suggests that bioinformatics predictions are useful in assessing the effects of newly discovered mutations on enzyme function, but care should be taken in extending predictions to the clinical phenotype especially in cases of heterozygosity. It also raises interesting questions about a dominant negative effect of some GALE missense alleles and potential compensatory mechanisms occurring in people born with clinical chemistry measurements suggesting a diagnosis of galactosemia.
Introduction
Type III galactosemia (OMIM#230350) is an inherited metabolic disease caused by a deficiency in the activity of UDP-galactose 4′-epimerase (GALE, EC 5.1.3.2).1–3 This enzyme catalyses the inter-conversion of UDP-galactose and UDP-glucose, a reaction that is part of the Leloir pathway of galactose catabolism.4,5 Additionally GALE has a second role in maintaining the pools of UDP-galactose and UDP-glucose (along with the related metabolites, UDP-N-acetylgalactosamine and UDP-N-acetylglucosamine), which are precursors in the synthesis of the sugar moieties of glycoproteins and glycolipids. The enzyme is a member of the short-chain dehydrogenase (SDR) family utilising a bound NAD+ cofactor and in regards to type III galactosemia the majority of mutations are missense, present throughout its structure.3 There is a wide spectrum of symptoms associated with the disease ranging from increased concentrations of galactose metabolites without clinical effects in the mildest forms to irreversible organ damage leading to physical and cognitive disability in the most severe forms.1,6–8 Currently, the only treatment is the removal of galactose (and its precursors such as lactose) from the diet. This treatment is unsatisfactory since, for those with the most severe forms of the disease, it only delays and reduces the extent of organ damage but cannot prevent or reverse it.7Biochemically, disease-associated mutations tend to reduce the activity of the enzyme.9–12 There is an approximate correlation between the extent of impairment in the turnover number (kcat) and the severity of disease.10,12 The majority of mutations alter residues away from the active site and the loss of activity is most likely caused by altered protein stability rather than direct effects on the active site residues.9,10,12–17 Interestingly both increased and decreased stability can be associated with reduced activity and disease.17 GALE is a homodimer with one molecule of NAD+ bound per subunit.18 This NAD+ is critical for the enzyme's activity and, in some of the disease-associated variants, one effect of reduced affinity for NAD+ is altered stability.13,17,19,20
To date, relatively few disease-associated mutations have been reported and GALE deficiency is considered to be rare. The incidence is strongly population dependent where it is estimated to be 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6700 in African Americans but approximately 1:64800 in Americans of European descent.21 However, the benign (asymptomatic) nature of the milder forms of the disease may mean that there are a significant number of undetected cases in the general population. Here we report the identification of a previously unreported mutation in GALE, which was associated with type III galactosemia at birth along with the characterisation of this variant protein. We also demonstrate the strengths of using bioinformatics methods to predict the biochemical effects of this mutation.
:6700 in African Americans but approximately 1:64800 in Americans of European descent.21 However, the benign (asymptomatic) nature of the milder forms of the disease may mean that there are a significant number of undetected cases in the general population. Here we report the identification of a previously unreported mutation in GALE, which was associated with type III galactosemia at birth along with the characterisation of this variant protein. We also demonstrate the strengths of using bioinformatics methods to predict the biochemical effects of this mutation.
Materials and methods
Molecular genetics
GALE mutation analysis was carried out by PCR amplification of the coding exons and adjacent intron regions (RefSeq NM_000403.3) followed by bidirectional Sanger sequencing using standard methods. The genetic diagnostic analyses were performed with full written informed consent in compliance with the relevant laws and institutional guidelines.Bioinformatics and molecular modelling
The crystal structures of human GALE with UDP-glucose and UDP-N-acetylglucosamine bound (PDB: 1EK5 and 1HZJ18,22) were energy minimised and solvated using YASARA.23 PyMol (http://www.pymol.com) was used to alter residue 89 to valine in both structures and the resulting variant structures re-minimised using YASARA. The minimised models are presented as ESI data† to this paper and were used in subsequent analyses. Accessible surface areas were calculated using GETAREA (http://curie.utmb.edu/getarea.html)24 and root mean squared deviations (RMSD) using the align function in PyMol. Multiple sequence alignments were calculated using ClustalW25 using reviewed GALE sequences in UniProt (http://www.uniprot.com). Conservation scores were obtained using the Scorecons server (http://www.ebi.ac.uk/thornton-srv/databases/cgi-bin/valdar/scorecons_server.pl)26 and SIFT tolerance scores from http://sift.jcvi.org/.27 Instability and aggregation tendencies were estimated using SNP effect 4.0 (http://snpeffect.switchlab.org/)28 and I-Mutant 3.0 (http://gpcr2.biocomp.unibo.it/cgi/predictors/I-Mutant3.0/I-Mutant3.0.cgi) at 37 °C and pH 8.8 in order to match the conditions used in in vitro enzyme assays.29,30Expression, purification and kinetic analysis of wild-type and variant human GALE
Wild-type human GALE was expressed in, and purified from, Escherichia coli HMS174(DE3) as previously described.10 The coding sequence was altered so that codon 89 encoded valine instead of alanine by site-directed mutagenesis using the QuikChange method.31 The variant protein was expressed and purified using the same protocol as the wild-type. Enzyme activity was measured in 50 mM Hepes–OH, pH 8.8 at 37 °C by coupling the production of UDP-glucose to its oxidation in an NAD+-requiring reaction catalysed by UDP-glucose dehydrogenase.10,32 Recombinant human UDP-glucose dehydrogenase was produced as previously described.16 Protein concentrations were estimated by the method of Bradford using BSA as a standard.33Analytical methods
Limited proteolysis was carried out using trypsin (10–900 nM supplemented with 2 mM calcium chloride) and GALE (16 μM) in a total volume of 10 μl. UDP-galactose (2 mM) was added as required. Reaction mixes were pre-incubated at 37 °C for 5 min prior to addition of the protease. They were then incubated for a further 30 min at 37 °C before being stopped by the addition of an equal volume of SDS-PAGE loading buffer (125 mM Tris–HCl, pH 6.8, 4% (w/v) SDS, 20% (v/v) glycerol, 1% (w/v) dithiothreitol, 0.002% (w/v) bromophenol blue) and heating at 95 °C for approximately 3 min. Reactions were analysed by 10% SDS-PAGE.Crosslinking of GALE (10 μM) with bis(sulphosuccinimidyl)suberate (BS3, 0.1–800 μM) was carried out at 37 °C in a reaction volume of 10 μl. Reaction mixes were pre-incubated for 5 min prior to the addition of crosslinker. Reactions were allowed to proceed for another 30 min at 37 °C before being stopped by addition of an equal volume of SDS-PAGE loading buffer and heating at 95 °C for approximately 3 min. Reactions were analysed by 10% SDS-PAGE.
Melting temperatures (Tm) of GALE (5 μM) were estimated (in triplicate) by differential scanning fluorimetry (DSF) using Sypro Orange (Sigma) as previously described.17,34 Fluorescence spectra of GALE (200 μl of 20 μM) were collected in triplicate using a Spectra Max Gemini X UV plate reader using an excitation wavelength of 280 nm.
Results and discussion
Clinical chemistry and molecular genetics
The female patient was tested for blood galactose in the neonatal period. An initial reading of 34.2 mg dl−1 was recorded (normal values < 20 mg dl−1). Eight days later this had dropped slightly to 31.2 mg dl−1. At six weeks, galactose 1-phosphate levels were 6.3 mg/100 ml erythrocytes (normal < 1.5 mg/100 ml) and GALE activity was 0.41 μmol h−1 ml−1 erythrocytes (normal 2.72–7.91 μmol h−1 ml−1 erythrocytes). Galactokinase and galactose 1-phosphate uridylyltransferase activity were both in the normal ranges. By eight months, galactose levels were 1.3 mg dl−1 and galactose 1-phoshate levels were 0.98 mg dl−1 (both within the normal range). At 21 months, galactose 1-phosphate had dropped further to 0.75 mg dl−1. The patient is developing normally without any treatment or dietary intervention.DNA sequencing revealed a heterozygous GALE variant c.266C>T (rs201437329) which is predicted to cause replacement of the amino acid alanine at residue 89 with valine, denoted p.Ala89Val. Ala-89 is located in the NAD+ binding site (Fig. 1a). The mutation has been registered with allele frequencies of 0.4% (29/6604) and 0.01% (8/65860), respectively, in Finnish and non-Finnish European individuals (ExAC database accessed 23rd Sept 2015, http://exac.broadinstitute.org) but to our knowledge has not yet been reported in a patient with GALE deficiency. No other alteration of potential functional relevance in the gene was identified.
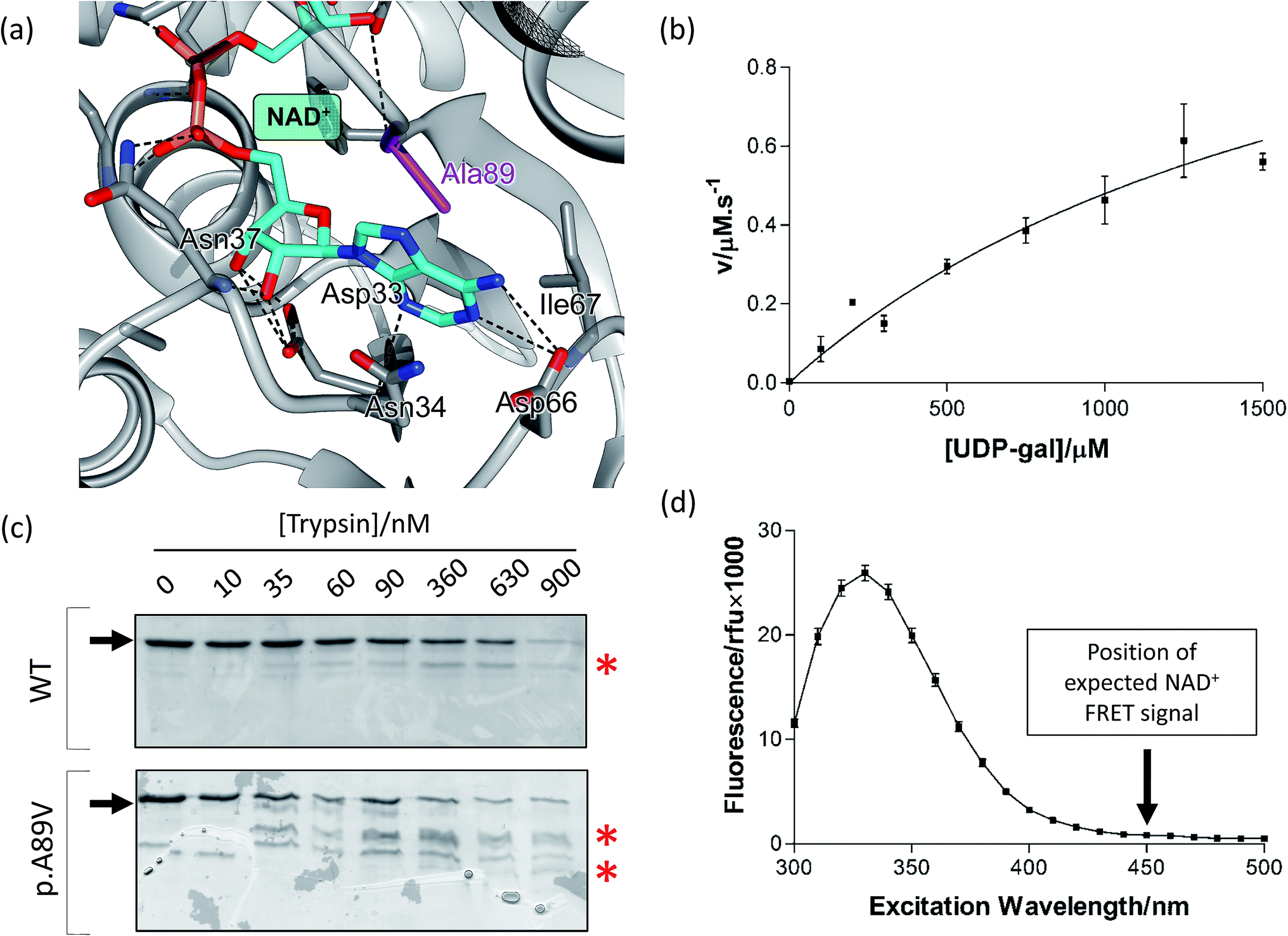 | ||
| Fig. 1 Biochemical analysis of GALE p.A89V. (a) Ala89 is located in the NAD+ binding site of the protein, close to the adenine moiety. Grey ribbon, backbone; grey sticks, key residues forming hydrogen bonds with NAD+; purple, Ala89; cyan, NAD+; black dashed lines, predicted hydrogen bonds. The image was generated in Chimera using PDB file 1EK5. (b) p.A89V (7 μM) has impaired kinetics compared to wild-type. Each point represents the mean of three determinations of the rate and the error bars the standard deviations of these means. The line represents a non-linear fit to the Michaelis–Menten equation. (c) p.A89V (16 μM) has reduced stability towards proteolytic digestion compared to the wild type. Results were analysed by 10% SDS-PAGE and the sizes of molecular mass markers are shown to the left of the gel in kDa. Red asterisks indicate degradation fragments for WT and p.A89V GALE. Arrows indicate full-length GALE protein. (d) Detection of NAD+ binding in p.A89V (20 μM) by FRET. The lack of signal at 450 nm indicates there is no NAD+ cofactor bound. Each point represents the mean of three determinations and the error bar the standard deviation of those means. | ||
Bioinformatics and molecular modelling
Multiple sequence alignment showed that Ala89 is highly conserved across GALE orthologues (ESI Fig. S1†). Indeed the conservation score26 was 0.99 and the SIFT tolerance score27 was 0.0 indicating a highly conserved residue in a site that is intolerant to this relatively conservative change. Molecular modelling predicted no significant change in the overall structures (RMSD of 0.297 Å and 0.345 Å for the UDP-glucose and UDP-N-acetylglucosamine bound structures respectively) and the accessible surface area of residue 89 was also similar (14.0–15.6%) for both WT and p.A89V bound to both product molecules (ESI Table S1†). Ala89 is located in the NAD+ cofactor-binding site, adjacent to the adenine moiety and the alteration of this residue to valine introduces a larger side-chain that results in a steric clash with the adenine moiety (Fig. 1a; ESI Fig. S2†). This indicates a potential to disrupt cofactor binding. Furthermore predictions with SNP effect 4.0 and I-Mutant 3.0 suggested that p.A89V is less stable than the wild-type, with an increased tendency to aggregate (ESI Table S2†). Collectively when compared to similar analyses carried out on variants for which the degree of enzymatic impairment is known,35 these results suggest that p.A89V is likely to be severely impaired compared to wild-type.Biochemical analysis of recombinant p.A89V
To gain insight into the biochemical effects of this mutation p.A89V was produced recombinantly and purified. The variant protein was substantially kinetically impaired compared to the wild-type enzyme (Fig. 1b) with a Michaelis constant (Km) increased almost 30-fold to 1900 ± 960 μM (compared to 69 μM for the wild-type10) and a turnover number (kcat) reduced 180-fold to 0.2 s−1. This is less than the value measured for the p.V94M variant (1.1 s−1; cf. the wild type value of 36 s−1) which is associated with severe forms of the disease.10Like the wild-type, p.A89V formed homodimers as judged by protein–protein crosslinking (data not shown), although there was a significant change in the protein's thermal stability (Tm of 48.8 ± 0.7 °C compared to 52.8 ± 0.3 °C for the wild-type under the same conditions; p = 0.0008, unpaired t-test).17 p.A89V was also more susceptible to proteolysis by trypsin (Fig. 1c) where the variant was degraded into more fragments and showed noticeable degradation at a lower protease concentrations. Both the wild-type and p.A89V were protected from this proteolysis in the presence of UDP-galactose (ESI Fig. S3†). Moreover in the wild-type protein, the bound NAD+ can be detected by FRET: when GALE is excited at 280 nm there is a large fluorescence emission peak at ∼340 nm (resulting from tryptophan and other aromatic residues) and a smaller peak at ∼450 nm (resulting from the bound NAD+).17 In p.A89V, the first peak was observed, but not the second (Fig. 1d). Therefore recombinant p.A89V, in contrast to the wild-type, likely does not contain appreciable amounts of NAD+. This suggests that the variant has reduced affinity for the cofactor, which is likely to be the main cause of the reduced stability towards proteases and the loss of activity compared to the wild-type. Therefore any activity that could be observed most likely results from the high concentrations of NAD+ (4 mM), which are necessarily present in the coupled enzyme assay.
Conclusions
Taken together, these results suggest that the ultimate cause of the initial GALE deficiency observed here results from the additional bulk of the valine side chain compared to that of alanine. This likely reduces the affinity for NAD+ substantially, and consequently the stability and activity of the protein. These biochemical results largely validate the predictive framework based on bioinformatic analyses that were previously proposed to assess newly discovered mutations.35 The patient initially presented with clinical chemistry measurements consistent with a diagnosis of type III galactosemia, but she remained asymptomatic and her metabolite and enzyme activity measurements are now in the normal range.Prolonged biochemical abnormalities and marked reduction in GALE activity are unusual in heterozygotes of GALE mutations. As such no relevant second alteration (on the other allele) was identified although a mutation in an intron, promoter or a large structural aberration involving parts of the gene cannot be excluded. Our analysis of the p.A89V mutation was not successful in determining the cause of the patient's apparent temporary galactosemic phenotype but it is possible that the raised galactose and galactose 1-phosphate levels in the period immediately after birth in this patient could be due to a dominant negative effect. The effects appear to have been more pronounced in the neonatal period and it is likely that the patient's milk intake decreased over the period in question that may provide a partial explanation although there may also be other, as yet unidentified, compensatory mechanisms at the cellular and molecular level. Nonetheless, in support of our hypothesis, dominant negative effects have been reported for the GALE mutations p.N34S and p.L183P when heterozygously expressed in a yeast model with the wild-type. These mutations resulted in 37% and 24% respectively of the activity of strains homozygous for wild-type GALE in contrast to the expected 50%.13 It also interesting to note that p.N34S affects a residue involved in NAD+ binding and p.L183P likely disturbs the overall structure of this region of the protein35 suggesting a link due to cofactor binding. Gaining insight into the structural and biochemical basis of this dominant negativity should be a priority for future research, especially as the majority of individuals with type III galactosemia are compound heterozygous.3 Such studies would give deeper insight into the role allelic heterogeneity plays as a whole in many metabolic disorders.
Acknowledgements
We thank Prof Aaron Maule (Institute for Global Food Security, Queen's University Belfast) for access to a qPCR machine used in DSF assays, and Dr Ursula Albrecht (University Children's Hospital Innsbruck, Austria) who cared for the patient and arranged the diagnostic work-up. TJM was funded by a Department of Employment and Learning, Northern Ireland (DELNI, UK) PhD studentship.Notes and references
- J. B. Holton, M. G. Gillett, R. MacFaul and R. Young, Arch. Dis. Child., 1981, 56, 885–887 CrossRef CAS PubMed.
- P. Maceratesi, N. Daude, B. Dallapiccola, G. Novelli, R. Allen, Y. Okano and J. Reichardt, Mol. Genet. Metab., 1998, 63, 26–30 CrossRef CAS PubMed.
- D. J. Timson, IUBMB Life, 2006, 58, 83–89 CrossRef CAS PubMed.
- L. F. Leloir, Arch. Biochem., 1951, 33, 186–190 CrossRef CAS PubMed.
- T. J. McCorvie and D. J. Timson, in Handbook of Glycosyltransferases and Related Genes, ed. N. Taniguchi, K. Honke, M. Fukuda, H. Narimatsu, Y. Yamaguchi and T. Angata, Springer, New York, 2014, vol. 2, ch. 133 Search PubMed.
- M. J. Henderson, J. B. Holton and R. MacFaul, J. Inherited Metab. Dis., 1983, 6, 17–20 CrossRef CAS PubMed.
- J. H. Walter, R. E. Roberts, G. T. Besley, J. E. Wraith, M. A. Cleary, J. B. Holton and R. MacFaul, Arch. Dis. Child., 1999, 80, 374–376 CrossRef CAS PubMed.
- J. L. Fridovich-Keil and J. H. Walter, in The Online Metabolic and Molecular Bases of Inherited Diseases, ed. D. Valle, A. L. Beaudet, B. Vogelstein, K. W. Kinzler, S. E. Antonarakis and A. Ballabio, McGraw-Hill, New York, 2008 Search PubMed.
- T. M. Wohlers and J. L. Fridovich-Keil, J. Inherited Metab. Dis., 2000, 23, 713–729 CrossRef CAS PubMed.
- D. J. Timson, FEBS J., 2005, 272, 6170–6177 CrossRef CAS PubMed.
- K. K. Openo, J. M. Schulz, C. A. Vargas, C. S. Orton, M. P. Epstein, R. E. Schnur, F. Scaglia, G. T. Berry, G. S. Gottesman, C. Ficicioglu, A. E. Slonim, R. J. Schroer, C. Yu, V. E. Rangel, J. Keenan, K. Lamance and J. L. Fridovich-Keil, Am. J. Hum. Genet., 2006, 78, 89–102 CrossRef CAS PubMed.
- J. S. Chhay, C. A. Vargas, T. J. McCorvie, J. L. Fridovich-Keil and D. J. Timson, J. Inherited Metab. Dis., 2008, 31, 108–116 CrossRef CAS PubMed.
- B. B. Quimby, A. Alano, S. Almashanu, A. M. DeSandro, T. M. Cowan and J. L. Fridovich-Keil, Am. J. Hum. Genet., 1997, 61, 590–598 CrossRef CAS PubMed.
- Y. L. Bang, T. T. Nguyen, T. T. Trinh, Y. J. Kim, J. Song and Y. H. Song, FEBS J., 2009, 276, 1952–1961 CrossRef CAS PubMed.
- A. L. Pey, E. Padin-Gonzalez, N. Mesa-Torres and D. J. Timson, Arch. Biochem. Biophys., 2014, 562, 103–114 CrossRef CAS PubMed.
- T. J. McCorvie, J. Wasilenko, Y. Liu, J. L. Fridovich-Keil and D. J. Timson, Biochimie, 2011, 93, 1747–1754 CrossRef CAS PubMed.
- T. J. McCorvie, Y. Liu, A. Frazer, T. J. Gleason, J. L. Fridovich-Keil and D. J. Timson, Biochim. Biophys. Acta, 2012, 1822, 1516–1526 CrossRef CAS PubMed.
- J. B. Thoden, T. M. Wohlers, J. L. Fridovich-Keil and H. M. Holden, Biochemistry, 2000, 39, 5691–5701 CrossRef CAS PubMed.
- U. S. Maitra and H. Ankel, Proc. Natl. Acad. Sci. U. S. A., 1971, 68, 2660–2663 CrossRef CAS.
- S. S. Wong and P. A. Frey, Biochemistry, 1977, 16, 298–305 CrossRef CAS PubMed.
- A. Alano, S. Almashanu, P. Maceratesi, J. Reichardt, S. Panny and T. M. Cowan, J. Invest. Med., 1997, 45, 191A Search PubMed.
- J. B. Thoden, T. M. Wohlers, J. L. Fridovich-Keil and H. M. Holden, J. Biol. Chem., 2001, 276, 15131–15136 CrossRef CAS PubMed.
- E. Krieger, K. Joo, J. Lee, J. Lee, S. Raman, J. Thompson, M. Tyka, D. Baker and K. Karplus, Proteins, 2009, 77(9), 114–122 CrossRef CAS PubMed.
- R. Fraczkiewicz and W. Braun, J. Comput. Chem., 1998, 19, 319–333 CrossRef CAS.
- M. A. Larkin, G. Blackshields, N. P. Brown, R. Chenna, P. A. McGettigan, H. McWilliam, F. Valentin, I. M. Wallace, A. Wilm, R. Lopez, J. D. Thompson, T. J. Gibson and D. G. Higgins, Bioinformatics, 2007, 23, 2947–2948 CrossRef CAS PubMed.
- W. S. Valdar, Proteins, 2002, 48, 227–241 CrossRef CAS PubMed.
- P. Kumar, S. Henikoff and P. C. Ng, Nat. Protoc., 2009, 4, 1073–1081 CrossRef CAS PubMed.
- G. De Baets, J. Van Durme, J. Reumers, S. Maurer-Stroh, P. Vanhee, J. Dopazo, J. Schymkowitz and F. Rousseau, Nucleic Acids Res., 2012, 40, D935–D939 CrossRef CAS PubMed.
- E. Capriotti, P. Fariselli and R. Casadio, Nucleic Acids Res., 2005, 33, W306–W310 CrossRef CAS PubMed.
- E. Capriotti, P. Fariselli, I. Rossi and R. Casadio, BMC Bioinf., 2008, 9(2), S6 CrossRef PubMed.
- W. Wang and B. A. Malcolm, BioTechniques, 1999, 26, 680–682 CAS.
- W. G. Ng, G. N. Donnell, J. E. Hodgman and W. R. Bergren, Nature, 1967, 214, 283–284 CrossRef CAS PubMed.
- M. M. Bradford, Anal. Biochem., 1976, 72, 248–254 CrossRef CAS PubMed.
- U. B. Ericsson, B. M. Hallberg, G. T. Detitta, N. Dekker and P. Nordlund, Anal. Biochem., 2006, 357, 289–298 CrossRef CAS PubMed.
- T. J. McCorvie and D. J. Timson, Gene, 2013, 524, 95–104 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra00306k |
| ‡ Current address: Structural Genomics Consortium, University of Oxford, Oxford OX3 7DQ, UK. |
| This journal is © The Royal Society of Chemistry 2016 |