
Bimetallic Schiff base complexes for stereoselective polymerisation of racemic-lactide and copolymerisation of racemic-lactide with ε-caprolactone†
Abstract
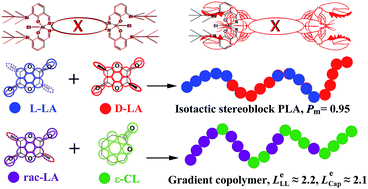
Three pairs of bimetallic Schiff base aluminum complexes (1a–6a) with different tetraamine backbone bridging parts and bulky silyl-substituted moieties on salicylidene were synthesized. Using these complexes as catalysts for ring opening polymerisation (ROP) of racemic-lactide (rac-LA), systematical stereoselectivity and kinetic studies on ROP of rac-LA were carried out. The Gibbs free energy difference between homo-propagation and cross-propagation (ΔG≠) calculated by activation entropy and enthalpy showed that bulky substituents at the 3-position enhanced the stereoselectivity. And the kinetic study results indicated that bulkier substituents with more steric hindrance reduced the ROP reactivity. Moreover, these complexes could also be used for copolymerisation of rac-LA and ε-caprolactone (ε-CL). Copolymers of poly(LA-co-CL) with various monomer ratios were synthesized. Kinetic research on copolymerisation as well as microstructure analysis demonstrated that poly (LA-co-CL) achieved by these complexes were tapered block copolymers.
 Please wait while we load your content...
Please wait while we load your content...

