
Exploiting photooxygenations mediated by porphyrinoid photocatalysts under continuous flow conditions†
Kleber T. de Oliveira*ab,
L. Zane Millera and
D. Tyler McQuade*a
aFlorida State University, Department of Chemistry and Biochemistry, Tallahassee, FL 32306-4390, USA. E-mail: tylermcquade@gmail.com
bUniversidade Federal de São Carlos, Departamento de Química, 13565-905, São Carlos, SP, Brazil. E-mail: kleber.oliveira@ufscar.br; Web: http://www.lqbo.ufscar.br
First published on 26th January 2016
Abstract
Photooxygenation reactions are a powerful synthetic tool to produce oxidized organic compounds; however, these reactions often exhibit experimental limitations including the production of complex mixtures that hinder desired product isolation and scale-up. Herein, we present a photocatalysed protocol under continuous flow conditions using a simple home built photoreactor and porphyrinoids as photocatalysts. Reaction conditions, long-term experiments, and scope demonstrate a protocol that is cost-effective, safe, reproducible and robust, thus allowing the production of relevant substituted naphthoquinones with interest in natural product synthesis and biological activity.
Introduction
Photooxygenation reactions have an important role in synthetic chemistry due to their high atom-economy and low cost.1 Over the last five decades researchers have recognized that singlet oxygen is the key excited intermediate for generating oxygenated compounds such as hydroperoxides,2 peroxides,3 dioxetanes,4 endoperoxides5 and sulfoxides.6The generation of singlet oxygen using photosensitizers is well-known.7 While many photosensitizers have been described,8 porphyrin derivatives are one of the most efficient classes of compounds for this purpose as supported by recent and relevant applications in organic synthesis.5,9
Since their discovery, photooxygenation processes have presented restrictions for their use in large scale reactors due to limitations imposed by the potential to form explosive intermediates, thus requiring the use of high dilutions and small scales. However, this limitation has recently been addressed by utilization of flow-based approaches to carry out photochemical transformations.10 Current advances in chemical synthesis under continuous flow conditions have in fact changed the way in which reactions are developed and scaled up in the pharmaceutical industry and research laboratories.11 Numerous advantages have been provided such as controlled mixing, fast heat transfer, control of residence time and process automation.12 Particularly, photochemical and photocatalysed transformations accelerated under continuous flow are one of the most prominent processes since significant improvements have been accomplished at the micro- and meso-scale, and successful reaction classes improved using these techniques due to the high efficiency of light irradiation and enhanced safety.10
Regarding the photooxygenations of naphthols, previous work performed in batch conditions has been published,13 as well as preliminary versions of photochemical flow devices. The pioneering work of Oelgemöller14 and co-workers showed the possibility to use both plug-flow and parallel falling films for photooxygenations under different continuous flow conditions. However, both methodologies and devices of these previous work displayed limitations and required many reaction cycles, high concentrations of photosensitizers (up to 5 mol%) with low conversions in some cases,14c inspiring us to explore improved and cost-competitive conditions for this continuous flow photooxygenation.
Herein, we have built and applied a simple, yet effective in-house engineered photoreactor to perform a comprehensive study of the photooxygenation of naphthols (Scheme 1). Different porphyrinoid and phthalocyanine derivatives were evaluated as photosensitizers (0.1–0.5 mol%), achieving high efficiency. Scope, robustness, and most importantly, scalability with a 24 h extended experiment are presented under continuous flow conditions, which allowed the production of relevant substituted naphthoquinones of interest in natural product synthesis and with well-recognized biological activities.15
 | ||
| Scheme 1 Photocatalytic cycle and photosentizers for the photooxygenation of 1. | ||
Results and discussion
First, in order to elucidate the best conditions for these photooxygenations, we constructed the photoreactor shown in Fig. 1 and S1 (ESI†). We used a reflexive aluminium-plate adapted with connections for a lamp with fan cooling, and a glass cylinder to coil the PFA (perfluoroalkoxy) tubing (for details, Fig. S1†). For process optimization we selected 5-hydroxynaphthol (1) to yield juglone (2) (Scheme 1).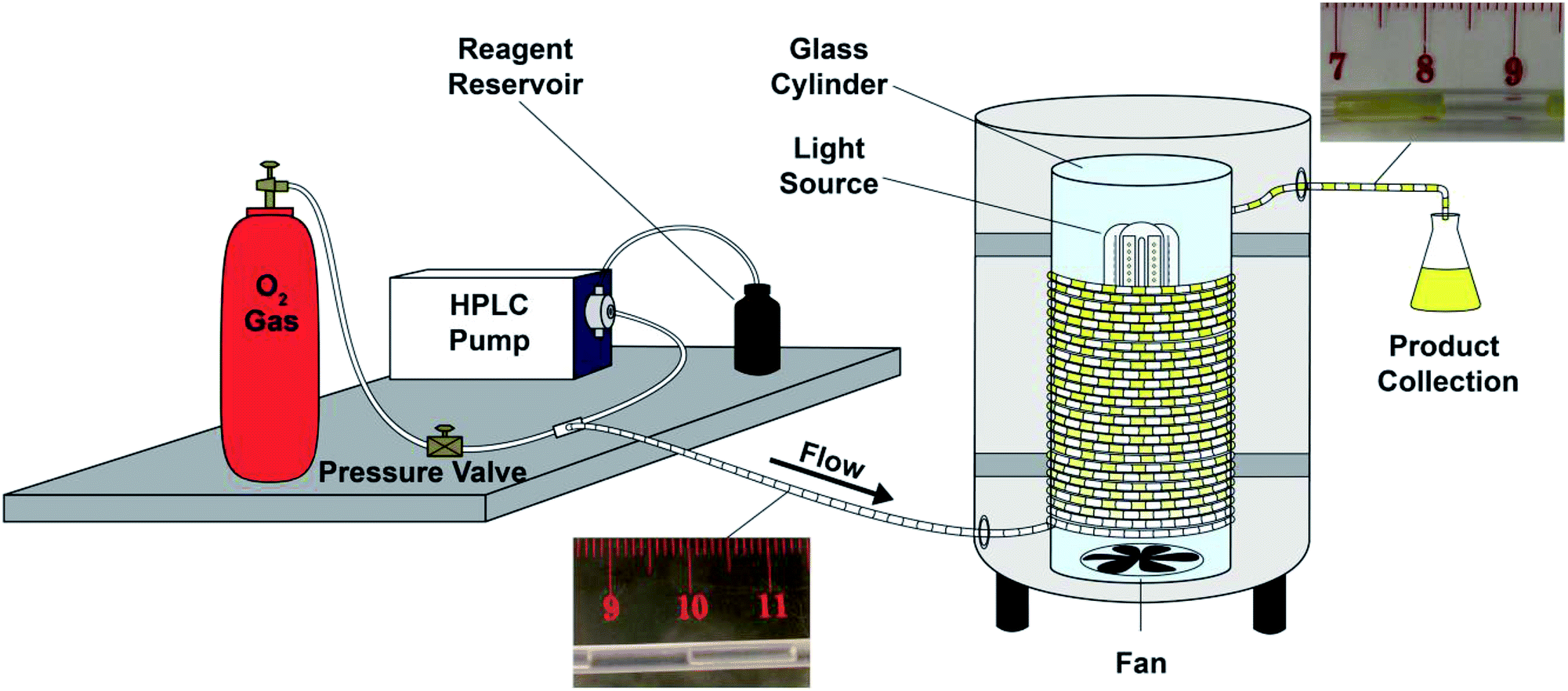 | ||
| Fig. 1 Flow chemistry setup for photooxygenations. | ||
We began the optimization by pumping of 100 mL of 3 mM solution of 1 with 0.1 mol% of the photocatalyst tetraphenylporphyrin (3) under three different plug-flow conditions (Table 1) in order to select the best flow rate and light source for the transformations (Scheme 1 and Fig. 1).
| Entry | Visible light source | Solution flow rate (mL min−1) | Juglone (2) yield (%) | Residence time (min) | STY (2)c (g per day) | ||
|---|---|---|---|---|---|---|---|
| 1st | 2nd | Avg | |||||
a Reactions performed by using 100 mL of a solution of the substrate 1 at 3.0 mmol L−1, photocatalyst 3 at 0.1 mol% in CH3CN![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CH2Cl2 (95:5), 1 cm plug-flow (regular oxygen and solution) in a 25 mL PFA tubing photoreactor (0.125 in (OD) × 0.065 in (ID)).b Isolated yield by column chromatography.c Considering the average yield (Avg). :CH2Cl2 (95:5), 1 cm plug-flow (regular oxygen and solution) in a 25 mL PFA tubing photoreactor (0.125 in (OD) × 0.065 in (ID)).b Isolated yield by column chromatography.c Considering the average yield (Avg). |
|||||||
| 1 | FLC, 45 W | 0.50 | 52 | 55 | 53 | 25.0 | 0.20 |
| 2 | FLC, 45 W | 0.75 | 58 | 60 | 59 | 16.7 | 0.33 |
| 3 | FLC, 45 W | 1.00 | 48 | 49 | 48 | 12.5 | 0.36 |
| 4 | LED, 24 W | 0.50 | 75 | 72 | 74 | 25.0 | 0.28 |
| 5 | LED, 24 W | 0.75 | 58 | 56 | 57 | 16.7 | 0.32 |
| 6 | LED, 24 W | 1.00 | 43 | 43 | 43 | 12.5 | 0.32 |
Using the FLC lamp (45 W) the best result was obtained with a flow rate of 0.75 mL min−1 (entry 2, Table 1) considering the yield, residence time, and space-time yield (STY). Similarly, the use of white LED lamp (24 W) and a flow rate of 0.75 mL min−1 (entry 5, Table 1) produced the best result in terms of yield, residence time and STY, and requiring approximately half of the energy compared to the FLC source (24 W vs. 45 W). The superiority of the white LED lamp can be explained by the comparison of the emission and absorption spectra (Fig. 2) for each light source and the porphyrin 3. Specifically, the broad emission band of white LED source (480–700 nm) encompasses the entire visible region of the absorption spectrum of 3 (500–670 nm).
 | ||
| Fig. 2 Comparison between the emission spectra of light sources and absorption spectra of 3. | ||
Thus, from the first six experiments it was possible to suggest the flow rate of 0.75 mL min−1 as the most adequate due to the productivity, and also because the most significant increase of STY was found in the range of 0.5 to 0.75 mL min−1. It is important to highlight that the reproducibility was also evaluated by performing each reaction twice (Table 1) with only minor variations of 1–3% yield, as observed in Table 1.
After establishing the best light source, different photocatalyst concentrations were examined from 0.1 to 0.5 mol% at three different flow rates (Table 2). From the results we conclude that 0.3 and 0.5 mol% are the best photosensitizer concentrations, but it was not clear if the use of 0.5 mol% would furnish the best cost-benefit.
| Entry | Solution flow rate (mL min−1) | Juglone (2) yield (%) using TPP (3) at 0.1 mol% | Juglone (2) yield (%) using TPP (3) at 0.2 mol% | Juglone (2) yield (%) using TPP (3) at 0.3 mol% | Juglone (2) yield (%) using TPP (3) at 0.5 mol% |
|---|---|---|---|---|---|
| a Reactions performed by using 100 mL of a solution of the substrate 1 at 3.0 mmL L−1, photocatalyst 3 in different concentrations in CH3CN:CH2Cl2 (95:5), 1 cm plug-flow (regular oxygen and solution) in a 25 mL PFA tubing photoreactor (0.125 in (OD) x 0.065 in (ID)). |
|||||
| 1 | 0.50 | 73 | 81 | 85 | 85 |
| 2 | 0.75 | 57 | 71 | 80 | 83 |
| 3 | 1.00 | 43 | 60 | 75 | 81 |
Subsequently, different concentrations of substrate 1 (Table 3) were assessed from 3.0 to 12.0 mM while maintaining the flow rate at 0.75 mL min−1 and employing two different photocatalyst concentrations (0.3 and 0.5 mol%). Comparing the experiments from entries 1–5 and 6–9, very similar yields and STY were observed between all comparable entries, however, the use of lower amounts of photocatalyst (0.3 mol%, entries 1–5, Table 3) were decisive for the choice of 0.3 mol% as the most ideal catalyst loading.
| Entry | Substrate 1 mmol L−1 | TPP (3) as photocatalyst (mol%) | Juglone (2) yield (%) | STY (2) (g per day) | Productivity mmol product per mmol catalyst per h |
|---|---|---|---|---|---|
| a Reactions performed by using 100 mL of a solution of the substrate 1, photocatalyst 3 at 0.3 mol% or 0.5 mol% in CH3CN:CH2Cl2 (95:5), solution flow rate (0.75 mL min−1), 1 cm plug-flow (regular oxygen and solution) in a 25 mL PFA tubing photoreactor (0.125 in (OD) × 0.065 in (ID)).b In these conditions it was observed a small amount of TPP (3) as a precipitate after the experiment with no serious blockage of the pump system. |
|||||
| 1 | 3.0 | 0.3 | 80 | 0.45 | 120 |
| 2 | 6.0 | 0.3 | 85 | 0.96 | 128 |
| 3 | 9.0 | 0.3 | 85 | 1.44 | 128 |
| 4 | 10.0 | 0.3 | 82 | 1.54 | 123 |
| 5b | 12.0 | 0.3 | 78 | 1.76 | 117 |
| 6 | 3.0 | 0.5 | 83 | 0.47 | 75 |
| 7 | 6.0 | 0.5 | 83 | 0.93 | 75 |
| 8b | 9.0 | 0.5 | 83 | 1.41 | 75 |
| 9b | 12.0 | 0.5 | 78 | 1.76 | 70 |
Another parameter which highlights this preference is the productivity of the photocatalyst which is almost two times greater for 0.3 mol% photocatalyst loading. The selected conditions to proceed with the methodology study are shown in entry 4, Table 3, since greater concentrations of substrate (up to 10 mM) required amounts of photocatalyst 3 (mol%) which caused precipitation to occur (entries 5, 8 and 9, Table 3). It is important to highlight that this optimized amount of TPP (3) (0.3 mol%, US dollars 33.1 mmol−1)16 is advantageous compared to similar photooxygenation procedures found in the literature14 using bengal rose (≥5 mol%, US dollars 40.4 mmol−1).16
After screening to find the best flow rate and substrate/photocatalyst concentrations we decided to evaluate different photocatalysts including additional porphyrinoids 4–6 and the phthalocyanine 7 (Table 4). Different photoactivities were observed most likely due to the different ability of these photosensitizers (3–7) to produce singlet oxygen since nearly the entire absorption spectra of 3–7 (Q-bands, 500–700 nm) were covered by the emission spectra of the LED lamp (430–700 nm). Clearly, the simplest and most cost-competitive photocatalyst 3 proved to be the most efficient giving us all of the optimized parameters required to advance the scope of this photooxygenation protocol. Before continuing with the scope of this protocol, we decided to showcase the robustness of this method and performed two 24 h experiments using the optimized conditions (substrate 1 at 10 mM, TPP 3 at 0.3 mol%, flow rate at 0.75 mL min−1 in CH3CN:CH2Cl2 95:5, and 1 cm plug-flow of oxygen and solution). Juglone (2) was obtained in both experiments in 72% (1.35 g) and 74% (1.40 g) yield, respectively, proving this protocol as reproducible and in agreement with STY (1.54 g) for these conditions (entry 4, Table 3).
| Entry | Photocatalysts at 0.3 mol% | Juglone (2) yield (%) | STY juglone (2) (g per day) | Productivity mmol product per mmol catalyst per h |
|---|---|---|---|---|
| a Reactions performed by using 100 mL of a solution of the substrate 1 at 10.0 mmol L−1, different photocatalysts at 0.3 mol% in CH3CN:CH2Cl2 (95:5), solution flow rate (0.75 mL min−1), 1 cm plug-flow (regular oxygen and solution) in a 25 mL PFA tubing photoreactor (0.125 in (OD) × 0.065 in (ID)). |
||||
| 1 | 3 | 82 | 1.54 | 123 |
| 2 | 4 | 15 | 0.28 | 22 |
| 3 | 5 | 72 | 1.35 | 108 |
| 4 | 6 | 6 | 0.11 | 9 |
| 5 | 7 | 18 | 0.34 | 27 |
In order to evaluate the scope, we selected different naphthol derivatives 8–17 containing various substituent groups in different positions (Table 5). The first general result is that naphthol derivatives containing electron-donating groups are invariably more reactive than the ones with electron-withdrawing groups, which is consistent with the accepted [4 + 2] pericyclic mechanism for this photooxygenation.1,13

| Entry | Substrate | Product and yield (%) under continuous flow conditionsb | Product and yield (%) in batch conditionsb,c |
|---|---|---|---|
| a All the reactions under continuous flow conditions were performed using the optimized conditions: LED lamp 24 W, 10 mM solutions of the substrates in CH3CN:CH2Cl2 (95:5) (100 mL), TPP (3) at 0.3 mol%, solution flow rate = 0.75 mL min−1, 1 cm plug-flow (oxygen-solution), residence time = 16.7 min, 25 mL photoreactor.b Isolated yields by using column chromatography.c Reactions performed using the residence time of the comparable continuous flow conditions (16.7 min).d An attempt to run this reaction under continuous flow conditions was performed, but due to the low solubility of the starting material it was not possible to finish it because the blockage of the system.e An attempt to perform this reaction in batch conditions was performed but no products were observed after 1 h.f Starting material totally recovered. |
|||
| 1 |  |
 |
 |
| 2 |  |
 |
 |
| 3 |  |
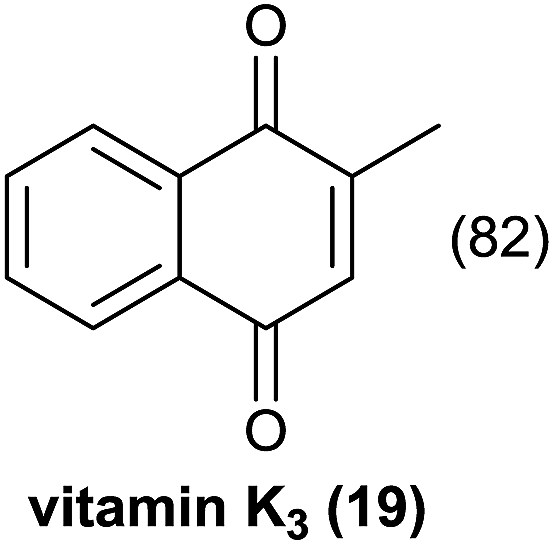 |
 |
| 4 | 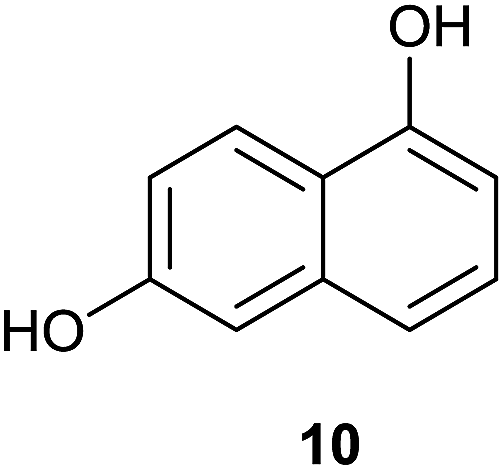 |
 |
 |
| 5 | 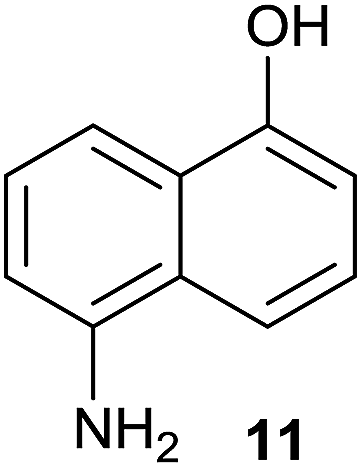 |
No reactiond | No reactione |
| 6 |  |
 |
 |
| 7 |  |
No reactionf | No reactionf |
| 8 | 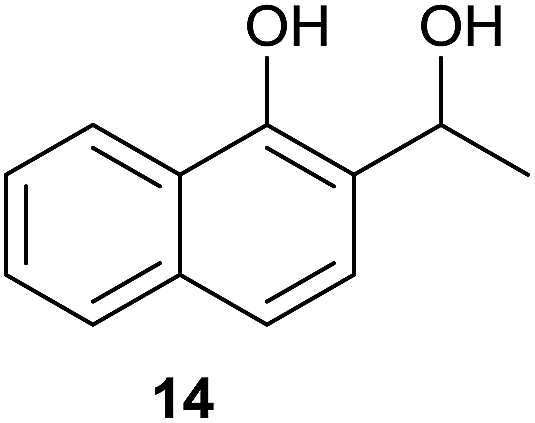 |
 |
 |
| 9 |  |
No reactionf | No reactionf |
| 10 | 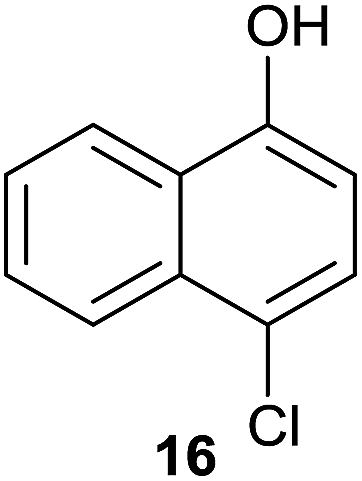 |
 |
 |
| 11 | 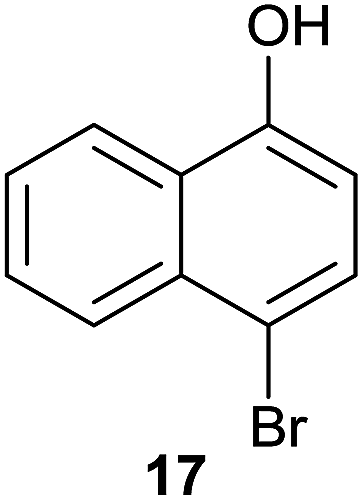 |
 |
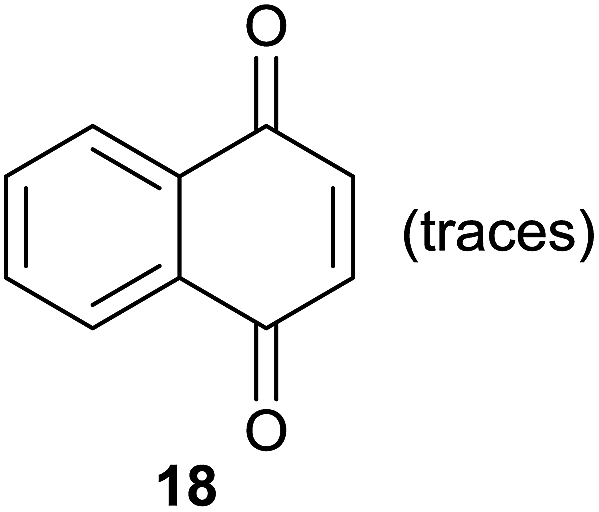 |
Comparing entries 1 and 3 (Table 5) the starting materials 1 and 8 yielded the expected corresponding naphthoquinones 2 and 19, respectively, with the same yield (82%) under continuous flow conditions, compared to 59% yield obtained for 18 (entry 2, Table 5). Also, the yields for batch conditions (using the same residence time, 16.7 min) were substantially lower than under continuous flow, providing a real advantage with the use of this photoreactor.
The activated 6-hydroxynaphthol (10) (entry 4, Table 5) yielded 20 in 46% yield under continuous flow conditions together with minor by-products which were difficult to separate. In an identical reaction performed using batch conditions only traces of 20 could be identified.
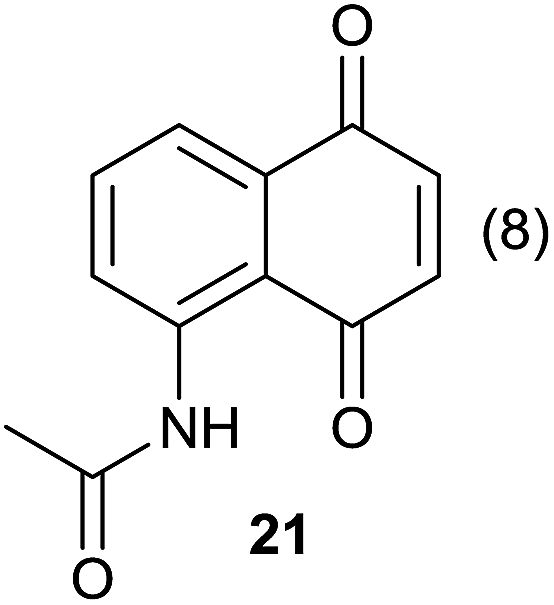
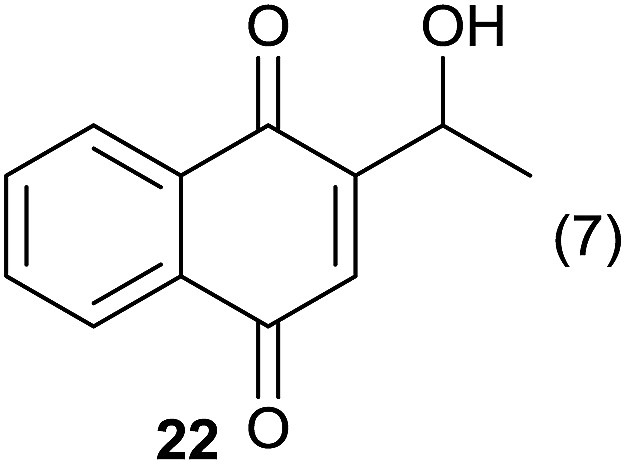
Compound 11 was submitted to photooxygenation under both flow and batch conditions, but unsuccessfully, most likely due to the very low solubility of this compound (entry 5, Table 5). On the other hand, the corresponding acetylated compound 12 yielded the corresponding naphthoquinone 21 in both flow (72% yield) and batch (8% yield) conditions (entry 6, Table 5). Similarly, the deactivated naphthol 13 (entry 7, Table 5) was unreactive in both flow and batch reactions, but the equivalent reduced compound 14 furnished the corresponding product 22 under both flow (75% yield) and batch (7% yield) conditions (entry 8, Table 5). The deactivated compound 15 also did not react (entry 9, Table 5), however, both compounds 16 and 17 yielded the corresponding naphthoquinone 18 (8 and 26% yield, respectively) under continuous flow conditions together with many by-products (entries 10 and 11, Table 5). It is important to highlight in entry 11 (Table 4) that we were able to isolate the brominated derivative 23 in 22% yield under continuous flow conditions, and we conclude that this was most likely possible via reaction of 18 and HOBr produced during the process.
Conclusions
In summary, we have developed an optimized protocol for photooxygenation of activated naphthols, screening some porphyrinoids as photocatalysts and many parameters under continuous flow conditions. In addition, we have shown the applicability and safety of this very simple device in process intensification by using mild and cost-competitive conditions to produce valuable compounds in gram-scale. Previous efforts described in the literature presented limitations requiring many reaction cycles due to the engineered devices or the photocatalysts which were used. Herein we have presented a simple and efficient solution for these problems allowing the production of relevant naphthoquinone derivatives in only one reaction cycle with short optimized residence time (16.7 min).Experimental section
All reagents and solvents were purchased from Aldrich or national US suppliers. When necessary, solvents and reagents were purified using standard procedures.17 Porphyrins 3–6 were obtained as described in the literature.7f,11r Phthalocyanine 7 was purchased from Sigma-Aldrich.1H and 13C NMR spectra were recorded with a Bruker Avance 600 spectrometer at 600 and 150 MHz, respectively. CDCl3 or DMSO-d6 was used as solvent and TMS (tetramethylsilane) as the internal reference. The chemical shifts are expressed in δ (ppm) and coupling constants (J) are given in Hertz (Hz). The UV-vis spectra were recorded with a Perkin-Elmer Lambda 25 spectrophotometer using 1 cm optical length quartz cuvettes at 25 °C and dichloromethane as solvent. Emission spectra of FLC and LED lamps were recorded an Ocean Optics Spectrometer HR2000CG-UV-NIR. MS analysis were performed using HP model 5973 mass selective detector with HP model 6890 + gas chromatograph; scanned from 50 to 550 amu; T = 70 °C for 3 min and then to 300 °C at 30 °C min−1.
All continuous flow experiments were carried out using a micro HPLC pump from ThalesNano and an in-house engineered photoreactor as specified in the ESI.† Analytical thin-layer chromatography was performed on glass plates (3 × 6 cm, 1 mm thick), Merck TLC silica-gel 60 F254.
General procedure for experiments under continuous flow conditions
A solution of naphthol (1 or 8–17) (0.3–1.2 mmol) was prepared in 10 mL of acetonitrile and sonicated for 2 min. After this was completed, 85 mL of oxygenated acetonitrile (previously bubbled with oxygen for 10 min) was added, and a solution of photocatalyst (0.1–0.5 mol%) in 5 mL of CH2Cl2 was added last. The reaction mixture was protected from light with an aluminium foil and the solution pumped into the photoreactor in different flow rates, as specified. The plug-flow was performed using a commercial oxygen cylinder adapted with a manometer (0–250 bar) and an intermediate valve for the fine adjustment of oxygen pressure (1.5–2.0 bar depending on the solution flow). All reactions were started only after previous stabilization of the plug-flow (ca. 1 cm each) using the desired flow rate of acetonitrile and oxygen. The product was also protected from external light and collected until the end of pumping process, when pure acetonitrile was used to clean the photoreactor maintaining the same initial flow rate. After recovering all of the reaction mixture, acetonitrile was distilled off under vacuum followed by purification by simple silica-gel plug filtration using CH2Cl2 as eluent or mixtures of solvents as specified in each example. In general, 50–70% of the photocatalyst was recovered, and the main product was easily isolated after the solvent evaporation.General procedure for experiments under batch conditions
A solution of naphthol (1 or 8–17) (1.0 mmol) was prepared in 10 mL of acetonitrile and sonicated for 2 min. After this was completed, 85 mL of oxygenated acetonitrile (previously bubbled with oxygen for 10 min) was added, and a solution of photocatalyst (0.3 mol%) in 5 mL of CH2Cl2 was added last. The reactions were carried out in a 250 mL Pyrex® round-bottom flask using the same LED lamp (24 W) from the flow photoreactor kept as close as possible to the round-bottom flask (see Fig. S3 – ESI†). A slow magnetic stirring and oxygen bubbling was used and the reaction maintained under irradiation for 16.7 min (the same residence time for comparison – Table 5). After the reaction, acetonitrile was distilled off under vacuum followed by purification by simple silica-gel plug filtration using CH2Cl2 as eluent or mixtures of solvents as specified in each example. When obtained, the main product was easily isolated after the solvent evaporation.General data and yields reported for results from Table 5
:hexanes 6:4 as eluent, Rf = 0.38, 59% yield (94.0 mg, 0.594 mmol). 1H NMR (600 MHz, CDCl3) δ (ppm): 6.99 (s, 2H); 7.75–7.79 (m, 2H); 8.08–8.12 (m, 2H). 13C NMR (150 MHz, CDCl3) δ (ppm): 126.4; 131.9; 133.9; 138.7; 185.0.:MeOH 9.5:0.5, Rf = 0.50, 46% yield (79.3 mg, 0.455 mmol). 1H NMR (600 MHz, DMSO-d6) δ (ppm): 6.97 (d, J = 10.2 Hz, 1H); 7.00 (d, J = 10.2 Hz, 1H); 7.18 (dd, J = 8.7 Hz, J = 2.5 Hz, 1H); 7.28 (d, J = 2.5 Hz, 1H); 7.86 (d, J = 8.7 Hz; 1H); 11.0 (br s, 1H). 13C NMR (150 MHz, DMOS-d6) δ (ppm): 111.6; 120.8; 123.7; 128.9; 133.7; 138.2; 139.0; 162.8; 183.6; 185.0.:ethyl acetate 6:4, Rf = 0.60, 72% yield (154.9 mg, 0.720 mmol). 1H NMR (600 MHz, CDCl3) δ (ppm): 2.31 (s, 3H); 6.93 (d, J = 10.2 Hz); 6.96 (d, J = 10.2 Hz, 1H); 7.74 (dd, J = 8.5 Hz, J = 1.1 Hz); 7.83 (dd, J = 7.5 Hz, J = 1.1 Hz, 1H); 9.09 (dd, J = 8.5 Hz, J = 1.1 Hz); 11.87 (br s, 1H). 13C NMR (150 MHz, CDCl3) δ (ppm): 25.6; 116.0; 121.9; 126.0; 132.2; 135.7; 138.0; 139.9; 141.3; 169.9; 184.4; 189.1.:ethyl acetate 9.5:0.5, Rf = 0.30, 75% yield (152.1 mg, 0.752 mmol). 1H NMR (600 MHz, CDCl3) δ (ppm): 1.52 (d, J = 6.4 Hz, 3H); 2.49 (d, J = 4.9 Hz, 1H); 5.02 (ddq, J = 6.4 Hz, J = 4.9 Hz, J = 1.5 Hz, 1H); 7.01 (d, J = 1.5 Hz, 1H); 7.74–7.78 (m, 2H); 8.06–8.12 (m, 2H). 13C NMR (150 MHz, CDCl3) δ (ppm): 22.6; 65.3; 126.2; 126.5; 131.9; 132.2; 132.9; 133.8; 134.0; 152.7; 185.3; 185.6.Acknowledgements
The authors would like to thank (McQuade projects: Clinton Health Access Initiative, Corning Glass, Gates Foundation, and NSF (CHE-1152020)), and the São Paulo Research Foundation (FAPESP) for the financial support for K. T. de Oliveira (BPE fellowship 2014/25538-6) and laboratory founding in Brazil (2013/06532-4 and 2013/07276-1). Thalesnano is thanked for providing the micro HPLC pump used in this work. Thanks are also due to Prof. T. J. Brocksom (UFSCar) and R. O. M. A. de Souza (UFRJ) for helpful discussions and to MsC. P. B. Momo and Dr M. C. Donatoni for the porphyrin synthesis.References
- (a) A. Greer, Acc. Chem. Res., 2006, 39, 797 CrossRef CAS PubMed; (b) A. Pace and E. L. Clennan, Tetrahedron, 2005, 61, 6665 CrossRef.
- C. L. Hugelshofer and T. Magauer, J. Am. Chem. Soc., 2015, 137, 3807 CrossRef CAS PubMed.
- A. O. Terent'ev, D. A Borisov, V. A. Vil and V. M. Dembitsky, Beilstein J. Org. Chem., 2014, 10, 34 CrossRef PubMed.
- N. S. Lopes, A. M. Yoshitake, A. F. Silva, V. X. Oliveira Jr, L. S. Silva, A. A. S. Pinheiro and L. F. M. L. Ciscato, Chem. Biol. Drug Des., 2015, 86, 1373 CAS.
- (a) M. Balci, Chem. Rev., 1981, 81, 91 CrossRef CAS; (b) S. T. Staben, X. Linghu and F. D. Toste, J. Am. Chem. Soc., 2006, 128, 12658 CrossRef CAS PubMed; (c) K. Gilmore, D. l. Kopetzki, J. W. Lee, Z. Horváth, D. T. McQuade, A. Seidel-Morgenstern and P. H. Seeberger, Chem. Commun., 2014, 50, 12652 RSC; (d) L. Novkovic, M. Trmcic, M. Rodic, F. Bihelovic, M. Zlatar, R. Matovic and R. N. Saicic, RSC Adv., 2015, 5, 99577 RSC; (e) B. Pieber, T. Glasnov and C. O. Kappe, Chem.–Eur. J., 2015, 21, 4368 CrossRef CAS PubMed; (f) T. S. A. Heugebaert, C. V. Stevens and C. O. Kappe, ChemSusChem, 2015, 8, 1648 CrossRef CAS PubMed; (g) Z. Amara, J. F. B. Bellamy, R. Horvath, S. J. Miller, A. Beeby, A. Burgard, K. Rossen, M. Poliakoff and M. W. George, Nat. Chem., 2015, 7, 489 CrossRef CAS PubMed.
- J. Dad'ová, E. Svobodová, M. Sikorski, B. König and R. Cibulka, ChemCatChem, 2012, 4, 620 CrossRef.
- (a) A. F. Uchoa, K. T. de Oliveira, M. S. Baptista, A. J. Bortoluzzi, Y. Iamamoto and O. A. Serra, J. Org. Chem., 2011, 76, 8824 CrossRef CAS PubMed; (b) C. M. B. Carvalho, T. J. Brocksom and K. T. de Oliveira, Chem. Soc. Rev., 2013, 42, 3302 RSC; (c) C. M. B. Carvalho, M. A. Fujita, T. J. Brocksom and K. T. de Oliveira, Tetrahedron, 2013, 69, 9986 CrossRef CAS; (d) F. A. B. dos Santos, A. F. Uchoa, M. S. Baptista, Y. Iamamoto, O. A. Serra, T. J. Brocksom and K. T. de Oliveira, Dyes Pigm., 2013, 99, 402 CrossRef CAS; (e) J. C. J. M. D. S. Menezes, M. A. F. Faustino, K. T. de Oliveira, M. P. Uliana, V. F. Ferreira, S. Hackbarth, B. Röder, T. T. Tasso, T. Furuyama, N. Kobayashi, A. M. S. Silva, M. G. P. M. S. Neves and J. A. S. Cavaleiro, Chem.–Eur. J., 2014, 20, 13644 CrossRef CAS PubMed; (f) P. B. Momo, C. Pavani, M. S. Baptista, T. J. Brocksom and K. T. de Oliveira, Eur. J. Org. Chem., 2014, 4536 CrossRef; (g) M. P. Uliana, L. Pires, S. Pratavieira, T. J. Brocksom, K. T. de Oliveira, V. S. Bagnato and C. Kurachi, Photochem. Photobiol. Sci., 2014, 13, 1137 RSC.
- (a) M. C. De Rosa and R. J. Crutchley, Coord. Chem. Rev., 2002, 233–234, 351 CrossRef CAS; (b) T. Montagon, M. Tofi and G. Vassilikogiannakis, Acc. Chem. Res., 2008, 41, 1001 CrossRef PubMed.
- J. Park, D. Feng, S. Yuan and H. C. Zhou, Angew. Chem., Int. Ed., 2015, 54, 430 CAS.
- (a) M. Oelgemöller and O. Shvydkiv, Molecules, 2011, 16, 7522 CrossRef PubMed; (b) J. P. Knowles, L. D. Elliott and K. I. Booker-Milburn, Beilstein J. Org. Chem., 2012, 8, 2025 CrossRef CAS PubMed; (c) K. Gilmore and P. H. Seeberger, Chem. Rec., 2014, 14, 410 CrossRef CAS PubMed; (d) Y. Su, N. J. W. Straathof, V. Hessel and T. Noël, Chem.–Eur. J., 2014, 20, 10562 CrossRef CAS PubMed; (e) M. Hurevich, J. Kandasamy, B. M. Ponnappa, M. Collot, D. Kopetzki, D. T. McQuade and P. H. Seeberger, Org. Lett., 2014, 16, 1794 CrossRef CAS PubMed; (f) D. B. Ushakov, K. Gilmore, D. Kopetzki, D. T. McQuade and P. H. Seeberger, Angew. Chem., Int. Ed., 2014, 53, 557 CrossRef CAS PubMed.
- (a) S. Mascia, P. L. Heider, H. T. Zhang, R. Lakerveld, B. Benyahia, P. I. Barton, R. D. Braatz, C. L. Cooney, J. M. B. Evans, T. F. Jamison, K. F. Jensen, A. S. Myerson and B. L. Trout, Angew. Chem., Int. Ed., 2013, 52, 12359 CrossRef CAS PubMed; (b) J. C. Pastre, D. L. Browne and S. V. Ley, Chem. Soc. Rev., 2013, 42, 8801 RSC; (c) D. T. McQuade and P. H. Seeberger, J. Org. Chem., 2013, 78, 6384 CrossRef CAS PubMed; (d) D. Cantillo, O. de Frutos, J. A. Rincón, C. Mateos and C. O. Kappe, J. Org. Chem., 2014, 79, 8486 CrossRef CAS PubMed; (e) V. D. Pinho, B. Gutmann, L. S. M. Miranda, R. O. M. A. de Souza and C. O. Kappe, J. Org. Chem., 2014, 79, 1555 CrossRef CAS PubMed; (f) B. Gutmann and C. O. Kappe, Eur. Pharm. Rev., 2015, 20, 37 Search PubMed; (g) B. J. Deadman, D. L. Browne, I. R. Baxendale and S. V. Ley Chem, Technol. Eng., 2015, 38, 259 CrossRef CAS; (h) J. M. Hawkins, Nature, 2015, 520, 302 CrossRef CAS PubMed; (i) B. Gutmann, D. Cantillo and C. O. Kappe, Angew. Chem., Int. Ed., 2015, 54, 6688 CrossRef CAS PubMed; (j) M. Baumann and I. R. Baxendale, Beilstein J. Org. Chem., 2015, 11, 1194 CrossRef CAS PubMed; (k) D. O. Nogueira, S. P. de Souza, R. A. C. Leão, L. S. M. Miranda and R. O. M. A. de Souza, RSC Adv., 2015, 5, 20945 RSC; (l) I. I. Junior, E. C. Gaudino, K. Martina, G. Cravotto, R. Luque and R. O. M. A. de Souza, RSC Adv., 2014, 4, 45772 RSC; (m) F. G. Finelli, L. S. M. Miranda and R. O. M. A. de Souza, Chem. Commun., 2015, 51, 3708 RSC; (n) G. C. O. Silva, J. R. Correa, M. O. Rodrigues, H. G. O. Alvim, B. C. Guido, C. C. Gatto, K. A. Wanderley, M. Fioramonte, F. C. Gozzo, R. O. M. A. de Souza and B. A. D. Neto, RSC Adv., 2015, 5, 48506 RSC; (o) L. C. Alves, A. L. Desiderá, K. T. de Oliveira, S. Newton, S. V. Ley and T. J. Brocksom, Org. Biomol. Chem., 2015, 13, 7633 RSC; (p) S. Matthies, D. T. McQuade and P. H. Seeberger, Org. Lett., 2015, 17, 3670 CrossRef CAS PubMed; (q) C. A. Correia, K. Gilmore, D. T. McQuade and P. H. Seeberger, Angew. Chem., Int. Ed., 2015, 54, 4945 CrossRef CAS PubMed; (r) P. B. Momo, B. S. Bellete, T. J. Brocksom, R. O. M. A. de Souza and K. T. de Oliveira, RSC Adv., 2015, 5, 84350 RSC.
- (a) R. J. Ingham, C. Battilocchio, D. E. Fitzpatrick, E. Sliwinski, J. M. Hawkins and S. V. Ley, Angew. Chem., Int. Ed., 2015, 54, 144 CrossRef CAS PubMed; (b) S. V. Ley, D. E. Fitzpatrick, R. J. Ingham and R. M. Myers, Angew. Chem., Int. Ed., 2015, 54, 3449 CrossRef CAS PubMed; (c) S. V. Ley, D. E. Fitzpatrick, R. M. Myers, C. Battilocchio and R. J. Ingham, Angew. Chem., Int. Ed., 2015, 54, 10122 CrossRef CAS PubMed.
- (a) J. Griffiths, K.-Y. Chu and C. Hawkins, J. Chem. Soc., Chem. Commun., 1976, 676 RSC; (b) D. Murtinho, M. Pineiro, M. M. Pereira, A. M. d'. A. Rocha Gonsalves, L. G. Arnaut, M. G. Miguel and H. D. Burrows, J. Chem. Soc., Perkin Trans. 2, 2000, 2441 RSC.
- (a) M. Oelgemöller, N. Healy, L. de Oliveira, C. Jung and J. Mattay, Green Chem., 2006, 8, 831 RSC; (b) A. Yavorskyy, O. Shvydkiv, C. Limburg, K. Nolan, Y. M. C. Delauréc and M. Oelgemöller, Green Chem., 2012, 14, 888 RSC; (c) O. Shvydkiv, C. Limburg, K. Nolan and M. Oelgemöller, J. Flow Chem., 2012, 2, 52 CrossRef CAS.
- For general applications and syntheses see: (a) K. W. Wellington, RSC Adv., 2015, 5, 20309 RSC; (b) A. K. Jordão, M. D. Vargas, A. C. Pinto, F. C. da Silva and V. F. Ferreira, RSC Adv., 2015, 5, 67909 RSC; (c) V. K. Tandon and S. Kumar, Expert Opin. Ther. Pat., 2013, 23, 1087 CrossRef CAS PubMed; (d) C. Lisboa, V. G. Santos, B. G. Vaz, N. C. De Lucas, M. N. Eberlin and S. J. Garden, J. Org. Chem., 2011, 76, 5264 CrossRef CAS PubMed; for (S)-espicufolin synthesis, see: (e) L. F. Tietze, K. M. Gericke, R. R. Singidi and I. Schuberth, Org. Biomol. Chem., 2007, 5, 1191 RSC; for (±)-γ-indomycinone synthesis, see: (f) D.-S. Hsu, T. Matsumoto and K. Suzuki, Chem. Lett., 2006, 35, 1016 CrossRef CAS; for mensacarcin synthesis see: (g) L. F. Tietze, C. Güntner, K. M. Gericke, I. Schuberth and G. Bunkoczi, Eur. J. Org. Chem., 2005, 2459 CrossRef CAS. For (+)-nocardione A synthesis, see: (h) D. L. J. Clive, S. P. Fletcher and D. Liu, J. Org. Chem., 2004, 69, 3282 CrossRef CAS PubMed; for rac-Juglomycin A synthesis, see: (i) G. A. Kraus and P. Liu, Synth. Commun., 1996, 26, 4501 CrossRef CAS.
- http://www.sigmaaldrich.com, accessed in December 21 2015.
- W. L. F. Armarego and D. D. Perrin, in Purification of Laboratory Chemicals, Butterworth-Heinemann, Oxford, UK, 4th edn, 2000 Search PubMed.
- N. Adarsh, M. Shanmugasundaram, R. R. Avirah and D. Ramaiah, Chem.–Eur. J., 2012, 18, 12655 CrossRef CAS PubMed.
- O. A. Kholdeeva, O. V. Zalomaeva, A. B. Sorokin, I. D. Ivanchikova, C. D. Pina and M. Rossi, Catal. Today, 2007, 121, 58 CrossRef CAS.
- F. Ameer, I. R. Green, K. Krohn and M. Sitoza, Synth. Commun., 2007, 37, 3041 CrossRef CAS.
- E. E. Coyle, K. Joyce, K. Nolana and M. Oelgemöller, Green Chem., 2010, 12, 1544 RSC.
- Z.-Y. Lin, Y.-L. Chen, C.-S. Lee and C.-P. Chuang, Eur. J. Org. Chem., 2010, 3876 CrossRef CAS.
- M. Naresh, P. Swamy, M. A. Kumar, M. M. Reddy, K. Srujana and N. Narender, Tetrahedron Lett., 2014, 55, 3926 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Copy of 1H and 13C NMR spectra. See DOI: 10.1039/c6ra00285d |
| This journal is © The Royal Society of Chemistry 2016 |