
Palladium-catalyzed decarboxylative ortho-aroylation of N-acetyl-1,2,3,4-tetrahydroquinolines with α-oxoarylacetic acids†
Abstract
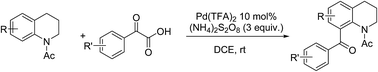
A mild, practical and efficient palladium-catalyzed decarboxylative ortho-aroylation of N-acetyl-1,2,3,4-tetrahydroquinolines with α-oxoarylacetic acids via C–H bond activation is described. This protocol provides efficient access to a series of C8-aroyl terahydroquinolines.
 Please wait while we load your content...
Please wait while we load your content...

