
Theoretical studies on oxidation-switchable second-order nonlinear optical responses of Metallosalen-Keggin polyoxometalate derivatives†
Teng-Ying Maab,
Ting Zhangb,
Bo Zhub,
Li-Kai Yan*b and
Zhong-Min Su*ab
aCollege of Chemistry, Jilin University, Changchun 130012, P. R. China
bInstitute of Functional Material Chemistry, Faculty of Chemistry, Northeast Normal University, Changchun 130024, P. R. China. E-mail: yanlk924@nenu.edu.cn; zmsu@nenu.edu.cn
First published on 25th May 2016
Abstract
To explore the effect of one-electron oxidation on nonlinear optical (NLO) responses, the frontier molecular orbitals, second-order NLO responses and electronic transition properties of three metallosalen–polyoxometalate compounds {SiW11O39[O(SiR)2M]}4− (R = (CH2)3N–CH(2-OPh); M = Pd, Ni and Co) (M-salen–POM) and their one-electron oxidized species were systematically investigated by density functional theory (DFT). Fukui function analysis shows that the M-salen is the oxidized center in M-salen–POM compounds. In addition, four functionals have been taken to confirm the switching behavior of the NLO properties by the one-electron oxidation. The βtot value of oxo-Pd-salen–POM is 98 times larger than that of Pd-salen–POM, indicating that the first hyperpolarizability is enhanced by one-electron oxidation. Time dependent (TD) DFT calculations show that the charge transfer from POM to M-salen, and intramolecular charge transfer within M-salen in oxidized species are beneficial to reduce the transition energy and improve the static first hyperpolarizabilities. The M-salen–POM may be a potential class of switchable nonlinear optical materials by one-electron oxidized processes.
Introduction
Design and synthesis of the second-order nonlinear optical (NLO) materials displaying large first hyperpolarizabilities (β) have been the focus of intensive investigations for several decades owing to their potential applications in the fields of storage, telecommunications, optical data processing and high speed optical switching.1–8 In the past few years, molecular switches exhibiting changes in color,9,10 luminescence,11,12 optical nonlinearity,13,14 or magnetic properties15–17 have been reported. Among them, switchable NLO materials have received much attention due to novel applications in electro-optic technologies.18,19 Several strategies have been proposed to achieve the effective switchable NLO materials, such as oxidation/reduction, protonation/deprotonation and photoisomerization, and so on.20–22 In this work, the oxidation was used to switch the NLO properties.Polyoxometalates (POMs) are a unique class of well-defined inorganic nanocluster compounds. POMs exhibit remarkable chemical and physical properties, which have been used in a variety of fields, such as medicine, catalysis, biology, analytical chemistry, materials science and so on.23–26 Over the past years, the POM-based organic–inorganic hybrid materials containing organic or organometallic moieties have especially received much attention, which exhibit remarkably large NLO response.27–29 POMs enable the formation of hybrid materials in which the delocalized electrons coexist in both the organic network and the inorganic clusters. Our groups have studied the geometric structures and the first hyperpolarizabilities of these kinds of hybrid materials by density functional theory (DFT).30,31
The modification of metal centers in organometallic compounds by changing the electronic and steric properties is an ongoing theme, which is important for controlling the reactivity of organometallic complexes. In previous report, a series of metallosalen compounds attached to a lacunary Keggin-type POM through an alkylene bridging spacer were synthesized by Neumann and co-workers.32 In these metallosalen–polyoxometalate (M-salen–POM) compounds, the POM exerts a significant intramolecular electronic effect on the metallosalen moiety leading to the formation of an oxidized metallosalen moiety. Meanwhile, the charge transfer between metallosalen donor and POM acceptor has been observed. Accompanying with the oxidization of metallosalen in these M-salen–POM compounds, many interesting phenomena might be discovered.
In this paper, three pairs of M-salen–POM (M = Pd, Ni and Co) compounds, including their one-electron oxidized states, were chosen to study the electronic and NLO properties. Herein, the M-salen–POMs, {SiW11O39[O(SiR)2M]}4− (R = (CH2)3N–CH(2-OPh); M = PdII, NiII and CoII) were named as Pd-salen–POM, Ni-salen–POM and Co-salen–POM, respectively, and the corresponding one-electron oxidized species (M = PdIII, NiIII and CoIII) were named as oxo-Pd-salen–POM, oxo-Ni-salen–POM and oxo-Co-salen–POM. The quantum chemical investigations on the structures, molecular orbitals, and first hyperpolarizabilities of the M-salen–POM compounds were performed to understand the effect of one-electron oxidation on NLO response. The results show that the oxidation of M-salen–POM could effectively reduce the transition energy and enhance the first hyperpolarizability. In other word, the NLO properties can be switched by one-electron oxidation in these compounds. It is expected that these M-salen–POM materials possessing switchable NLO properties could enrich NLO materials.
Computational details
The density functional theory (DFT) with Becke's three parameter exchange functional combined with the Lee–Yang–Parr correlation functional (B3LYP)33–35 has been widely used to optimize the molecular geometries. In fact, the B3LYP functional can well describe the geometric structures of POMs-based inorganic–organic hybrid compounds. The geometric parameters obtained by the B3LYP functional are in good agreement with the experimental values.36–38 Herein, the geometries of all compounds were optimized and characterized as energy minima by B3LYP functional. The solvent effect of N,N-dimethylformamide (DMF) was considered by the polarizable continuum model (PCM).39 The LANL2DZ basis set with Los Alamos relativistic effective core potentials (ECPs)40 was employed for metal atoms (W, Co, Ni, Pd), while the 6-31G* basis set was used for C, H, O, N and Si elements.The static first hyperpolarizability is termed as the zero frequency first hyperpolarizability, which is an estimate of the intrinsic molecular first hyperpolarizability in the absence of resonance effect. The first hyperpolarizability, βtot, for all compounds were calculated by using the following equation (eqn (1)):41
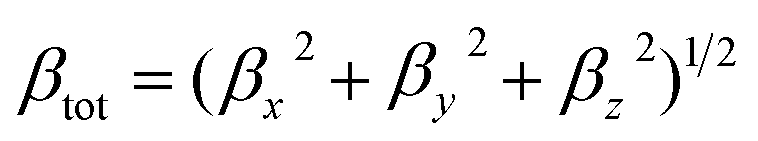 | (1) |
 | (2) |
The functionals for evaluating the first hyperpolarizabilities are usually general gradient approximation (GGA) in recent years. However, the Hartree–Fock (HF) exchange function and the long-range function must be included to obtain more reasonable results. Thus, several types of density functionals were selected to examine the reliability of the calculated results. The meta-GGA M06-2X functional42 includes a high percentage of HF exchange has been proved to be a good method to calculate the first hyperpolarizabilities.21,43 Also, the CAM-B3LYP44 and LC-BLYP45 including long range corrections were employed because these DFT functionals are excellent for computing the first hyperpolarizabilities of transition metal complexes.21,38,43,46,47
Moreover, in order to obtain a more intuitive description on NLO behaviors of compounds, time dependent (TD) DFT method was applied to determine the excitation energies of excited states at the PBE0 (ref. 48)/6-31G** level (LANL2DZ basis set for W, Co, Ni and Pd atoms) due to its efficiency and accuracy,37,49,50 and PCM was employed in TDDFT calculations. Furthermore, in order to better describe the connection of the first hyperpolarizabilities and the electronic transition of excited states, the PBE0 functional were selected to calculate the first hyperpolarizabilities. The open-shell calculations were performed using unrestricted methods. All calculations in this work were carried out by using the GAUSSIAN 09 program package.51
Results and discussion
Geometric and electronic structures
In this paper, the electronic configuration of ground state of systems Pd-salen–POM, oxo-Pd-salen–POM, Ni-salen–POM, oxo-Ni-salen–POM, Co-salen–POM and oxo-Co-salen–POM are singlet, doublet, singlet, doublet, quartet and triplet, respectively. The energies on different electronic configuration for the systems are listed in Table S1.† The geometrical structures of M-salen–POM compounds are similar and are sketched in Fig. 1. The comparison on structural parameters between computed results and experimental ones cannot be carried out as crystal structural parameters are not available. Furthermore, to analyze the effect of oxidation processes on the geometric structure, the selected bond lengths for three pairs of compounds obtained at B3LYP/6-31G* (LANL2DZ basis set for W, Co, Ni and Pd atoms) level are shown in Table 1. It can be seen that the differences on the calculated bond lengths between M-salen–POM compounds and their one-electron oxidized species mainly concentrate on the M-salen part. It suggests the oxidation may take place on M-salen. There is no doubt that the redox property of compound is closely relative to the nature of the frontier molecular orbitals. And the highest occupied molecular orbital (HOMO) reflects the nature of the oxidation of compounds. So the HOMOs of Pd-salen–POM, Ni-salen–POM and Co-salen–POM are analyzed. As shown in Fig. S1,† the HOMOs of studied M-salen–POMs are similar and entirely locate on the M-salen unit. It proposes that the M-salen unit will be the oxidized center in one-electron oxidized process. Combined with the prediction from the geometrical structures, it is reasonable that the oxidation process occurs on the M-salen part. | ||
| Fig. 1 The structure of calculation model. | ||
| Compounds | Pd-salen–POM | Oxo-Pd-salen–POM | Ni-salen–POM | Oxo-Ni-salen–POM | Co-salen–POM | Oxo-Co-salen–POM |
|---|---|---|---|---|---|---|
| M–O | 2.031 | 2.025 | 1.874 | 1.878 | 1.909 | 1.841 |
| M–N | 2.118 | 2.102 | 1.991 | 1.988 | 2.036 | 1.987 |
| O–C1 | 1.308 | 1.293 | 1.307 | 1.296 | 1.304 | 1.293 |
| N–C2 | 1.296 | 1.297 | 1.299 | 1.297 | 1.299 | 1.311 |
In addition, the Fukui function has been considered as a common descriptor of reaction activity in the corresponding system.52 Fukui function f(r) is defined as

| fA+ = qAN+1 − qAN (for nucleophilic attack) |
| fA− = qAN − qAN−1 (for electrophilic attack) |
where qA is the electronic population of atom A in a molecule.
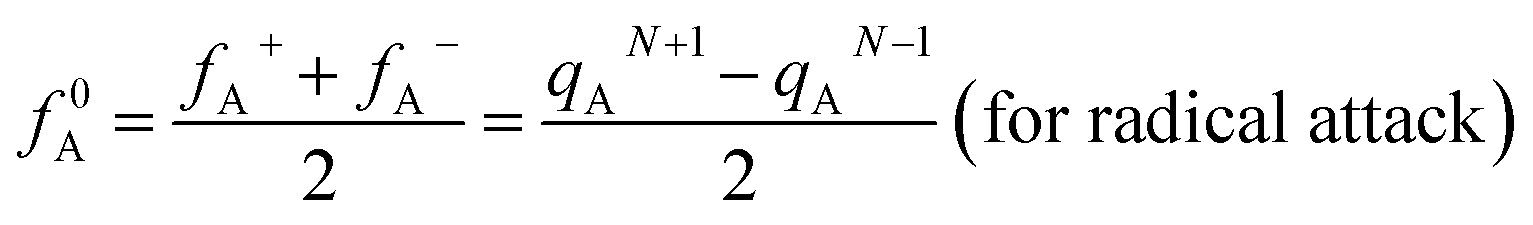
In order to further verify the oxidized center by an intuitive expression, the Fukui function was visualized with Multiwfn3.2.1 (ref. 53) and shown in Fig. 2. The oxidation process is discussed in this work, so the f− was analyzed here. The result is well agreement with the experimental phenomenon32 that the M-salen is the oxidized center in M-salen–POM (M = Pd, Ni and Co). Accompanying with the oxidized process, the electronic structure might be affected, which further affects the static first hyperpolarizabilities. The influences of oxidized process on the NLO response of compounds are investigated in the following sections.
 | ||
| Fig. 2 The isosurface of Pd-salen–POM, Ni-salen–POM and Co-salen–POM. | ||
Switchable second-order NLO properties
The static first hyperpolarizability (βtot) have been calculated by M06-2X, CAM-B3LYP, LC-BLYP and PBE0 functionals with the 6-31G** basis set (LANL2DZ basis set for metal atoms) in this work and the βtot values are shown in Table 2. The βtot values reveal a dependence on the functional. For example, the βtot value of oxo-Pd-salen–POM by LC-BLYP is twenty-one and fifteen percent of those by M06-2X and CAM-B3LYP, respectively. And the βtot value by PBE0 is larger than those of other three functionals. It seems that the LC-BLYP functional underestimates and the PBE0 overestimates the first hyperpolarizabilities for studied oxidized compounds in this work. Although the βtot values by the hybrid functionals M06-2X and PBE0 and the long range correction functionals CAM-B3LYP and LC-BLYP are different, the four functionals yield the same trend on βtot values. As can be seen in Table 2, the βtot values of Pd-salen–POM, Ni-salen–POM and Co-salen–POM are 3.59 × 10−30, 3.82 × 10−30 and 4.62 × 10−30 esu by M06-2X functional, and the difference on βtot value among three compounds is subtle. But the βtot values of their one-electron oxidized species are obviously increased. In order to investigate the influence of one-electron oxidation on the switching behavior of first hyperpolarizability, the βtot values of M-salen–POM and their corresponding one-electron oxidized species are compared. From the results in Table 2, the βtot value of oxo-Pd-salen–POM is 377.27 × 10−30 esu by the CAM-B3LYP functional, which is 98 times larger than that of Pd-salen–POM (3.85 × 10−30 esu). The other two pairs of compounds also show good switching properties on second-order NLO coefficients, and the multiplying factor is about 35 (β(oxo-Ni-salen–POM)/β(Ni-salen–POM)) and 218 (β(oxo-Co-salen–POM)/β(Co-salen–POM)) times by CAM-B3LYP functional, respectively. It proposes that the oxidized process tremendously enhances the static first hyperpolarizability. So these kinds of compounds might be promising candidates for switchable NLO materials.| Compounds | M06-2X | CAM-B3LYP | LC-BLYP | PBE0 |
|---|---|---|---|---|
| Pd-salen–POM | 3.59 | 3.85 | 5.84 | 8.12 |
| Oxo-Pd-salen–POM | 269.42 | 377.27 | 57.13 | 2293.41 |
| Ni-salen–POM | 3.82 | 4.02 | 5.73 | 8.98 |
| Oxo-Ni-salen–POM | 106.21 | 140.36 | 25.82 | 581.76 |
| Co-salen–POM | 4.62 | 10.10 | 7.63 | 6.30 |
| Oxo-Co-salen–POM | 535.64 | 2178.81 | 752.80 | 8495.04 |
TDDFT calculations
According to an approximately two-level expression proposed by Oudar,54 the βtot value is proportional to the oscillator strength (f) and the difference of dipole moment between ground state and crucial excited state (Δμ) and inversely proportional to the third power of transition energy (ΔE3). To provide the understanding about the oxidation effect on the first hyperpolarizabilities of M-salen–POM, we focus on the crucial excited states of these compounds. The crucial excited state is the lowest excited state with relatively large oscillator strength among all excited states. In this work, the ΔE, f, Δμ and λmax (maximum absorption wavelengths of the UV-vis spectra) were computed by PBE0 functional, and the results are listed in Tables S2† and 3. Firstly, the calculated λmax and the experimental one (Table S2†) are compared, and the difference is less than 30 nm, confirming that the PBE0 functional is exact for simulating the absorption spectra of these compounds. Furthermore, the ΔE value of oxo-Pd-salen–POM is 1.539 (2.655) eV, which is smaller than that of Pd-salen–POM (3.518 eV). Similarly, the ΔE values of oxo-Ni-salen–POM (2.010 eV) and oxo-Co-salen–POM (1.603 eV) are also smaller than those of Ni-salen–POM (3.641 eV) and Co-salen–POM (3.431 eV), respectively. The results show that the oxidization greatly reduce the ΔE value. Obviously, the order of ΔE3 is inversely proportional to that of the βtot values from the two-level expression. The Δμf/ΔE3 values of oxidized species are larger than their non-oxidized species, which result in the larger βtot values for oxidized species. The result show that the order of Δμf/ΔE3 values is in agreement with βtot values calculated with the PBE0 functional that the βtot values of oxidized species are larger than their non-oxidized species. Significantly, the transition energy is the decisive factor on determining the βtot values of these compounds.| Compounds | Pd-salen–POM | Ni-salen–POM | Co-salen–POM | Oxo-Pd-salen–POM | Oxo-Ni-salen–POM | Oxo-Co-salen–POM |
|---|---|---|---|---|---|---|
| Excited state | S24 | S33 | S60 | S4/S19 | S15 | S10 |
| ΔE (eV) | 3.518 | 3.641 | 3.431 | 1.539/2.655 | 2.010 | 1.603 |
| f | 0.148 | 0.200 | 0.027 | 0.027/0.035 | 0.049 | 0.106 |
| Δμ | 1.311 | 1.498 | 0.570 | 0.847/0.735 | 1.005 | 1.646 |
| Δμf/ΔE3 (10−3) | 4.456 | 6.207 | 0.381 | 6.274/1.375 | 6.188 | 42.358 |
Furthermore, we focused on analyzing the molecular orbitals of the crucial excited states for the three pairs of compounds to understand the origin of βtot values. As shown in Fig. 3 and Table S2,† the crucial excited states of Pd-salen–POM are mainly made up of H(HOMO) → L(LUMO)+21 (34%), H−1 → L+19 (31%) and H → L+7 (8%) electron transitions. The occupied molecular orbitals (H and H−1) entirely localize on the M-salen (the p orbitals of C, O and N atoms and the d orbitals of Pd atoms), and the unoccupied molecular orbitals (L+7, L+19 and L+21) mainly localize on the POM (the d orbitals of W atoms and the p orbitals of O atoms) and M-salen. Therefore, these electron transitions generate the mixing charge transfer from M-salen to POM and the intramolecular π → π* transition within M-salen, which is classified as type I charge transfer. The crucial excited states of the one-electron oxidized species oxo-Pd-salen–POM contain the βH−38 → βL (41%), αH−1 → αL+4 (12%) and αH−39 → αL+4 (8%) transitions in excited state S19 and βH−1 → βL (98%) transitions in excited state S3. Contrast to the Pd-salen–POM, the occupied molecular orbitals of oxo-Pd-salen–POM mainly localize on the M-salen and POM, and the unoccupied molecular orbitals entirely localize on M-salen, which is assigned the charge transfer from POM to the M-salen and the intramolecular charge transfer (π → π*) in M-salen, which is defined as type II charge transfer. It clearly shows that POM acts as the electron acceptor in type I charge transfer (Pd-salen–POM), while it acts as electron donor in type II charge transfer (oxo-Pd-salen–POM). As mentioned above, the βtot value of oxo-Pd-salen–POM is much larger than that of Pd-salen–POM. So the mixing transitions for the type II charge transfer might be helpful to enhance the first hyperpolarizability of oxo-Pd-salen–POM. Furthermore, the molecular orbitals of the crucial excited states for other two pairs of compounds are list in Fig. S2 and S3.† The results show that the charge transfers in Ni-salen–POM and Co-salen–POM are similar to that of Pd-salen–POM, which exhibits the type I charge transfer. And the charge transfers in oxo-Ni-salen–POM and oxo-Ni-salen–POM resemble to that of oxo-Pd-salen–POM, which belongs to the type II charge transfer.
 | ||
| Fig. 3 Molecular orbitals of Pd-salen–POM and oxo-Pd-salen–POM involved in the crucial excited states. | ||
Conclusions
In summary, three pairs of compounds were systematically investigated with DFT and TDDFT methods to explore the one-electron oxidation effect on the static first hyperpolarizability. Fukui function analysis shows that the M-salen is the oxidized center in M-salen–POM compounds. The one-electron oxidized process obviously enhances the static first hyperpolarizability. The TDDFT calculations exhibit that the type II charge transfer in one-electron oxidized species that POM acts as electron donor might play a key role for effectively reducing the transition energy and improving the static first hyperpolarizability. The βtot value of oxo-Pd-salen–POM is 377.27 × 10−30 esu, which is 98 times larger than that of Pd-salen–POM by CAM-B3LYP functional. So this kind of M-salen–POM compounds might be excellent switchable NLO material. The analysis of the effect of one-electron oxidation on their optical responses might provide important information of these compounds for switchable NLO applications.Acknowledgements
This work is supported by the National Natural Science Foundation of China (No. 21571031 and 21131001).Notes and references
- J. Zyss, Molecular Nonlinear Optics: Materials, Physics and Devices, Academic Press, Boston, 1994 Search PubMed.
- H. S. Nalwa and S. Miyata, Nonlinear Optics of Organic Molecules and Polymers, CRC Press, Boca Raton, Florida, 1997 Search PubMed.
- S. Di Bella, I. Fragalà, I. Ledoux, M. A. Diaz-Garcia and T. J. Marks, J. Am. Chem. Soc., 1997, 119, 9550 CrossRef CAS.
- J. P. Costes, J. F. Lamère, C. Lepetit, P. G. Lacroix and F. Dahan, Inorg. Chem., 2005, 44, 1973 CrossRef CAS PubMed.
- E. Cariati, M. Pizzotti, D. Roberto, F. Tessore and R. Ugo, Coord. Chem. Rev., 2006, 250, 1210 CrossRef CAS.
- R. L. Zhong, J. Zhang, S. Muhammad, Y. Y. Hu, H. L. Xu and Z. M. Su, Chem.–Eur. J., 2011, 17, 11773 CrossRef CAS PubMed.
- H. L. Xu, Z. R. Li, D. Wu, B. Q. Wang, Y. Li, F. L. Gu and Y. Aoki, J. Am. Chem. Soc., 2007, 129, 2967 CrossRef CAS PubMed.
- R. L. Zhong, H. L. Xu, S. Muhammad, J. Zhang and Z. M. Su, J. Mater. Chem., 2012, 22, 2196 RSC.
- M. Irie, Chem. Rev., 2000, 100, 1683 CrossRef CAS PubMed.
- V. De Waele, U. Schmidhammer, T. Mrozek, J. Daub and E. Riedle, J. Am. Chem. Soc., 2002, 124, 2438 CrossRef CAS PubMed.
- A. P. de Silva, H. Q. Nimal Gunaratne, T. Gunnlaugsson, A. J. M. Huxley, C. P. McCoy, J. T. Rademacher and T. E. Rice, Chem. Rev., 1997, 97, 1515 CrossRef CAS PubMed.
- S. J. Yoon, J. W. Chung, J. Gierschner, K. S. Kim, M. G. Choi, D. Kim and S. Y. Park, J. Am. Chem. Soc., 2010, 132, 13675 CrossRef CAS PubMed.
- K. A. Green, M. P. Cifuentes, M. Samoc and M. G. Humphrey, Coord. Chem. Rev., 2011, 255, 2530 CrossRef CAS.
- I. Asselberghs, K. Clays, A. Persoons, M. D. Ward and J. Mc Cleverty, Mater. Chem., 2004, 14, 2831 RSC.
- P. Gutlich, Y. Garcia and T. Woike, Coord. Chem. Rev., 2001, 219, 839 CrossRef.
- Z. P. Ni, X. M. Ren, J. Ma, J. L. Xie, C. L. Ni, Z. D. Chen and Q. J. Meng, J. Am. Chem. Soc., 2005, 127, 14330 CrossRef CAS PubMed.
- M. Z. Zhu, C. M. Aikens, M. P. Hendrich, R. Gupta, H. F. Qian, G. C. Schatz and R. C. Jin, J. Am. Chem. Soc., 2009, 131, 2490 CrossRef CAS PubMed.
- B. J. Coe, S. Houbrechts, I. Asselberghs and A. Persoons, Angew. Chem., Int. Ed., 1999, 38, 366 CrossRef CAS.
- B. J. Coe, Acc. Chem. Res., 2006, 39, 383 CrossRef CAS PubMed.
- N. N. Ma, S. L. Sun, C. G. Liu, X. X. Sun and Y. Q. Qiu, J. Phys. Chem. A, 2011, 115, 13564 CrossRef CAS PubMed.
- T. Y. Ma, N. N. Ma, L. K. Yan, T. Zhang and Z. M. Su, J. Phys. Chem. A, 2013, 117, 10783 CrossRef CAS PubMed.
- V. Aubert, V. Guerchais, E. Ishow, K. Hoang-Thi, I. Ledoux, K. Nakatani and H. Le Bozec, Angew. Chem., Int. Ed., 2008, 47, 577 CrossRef CAS PubMed.
- M. T. Pope and A. Müüller, Angew. Chem., Int. Ed. Engl., 1991, 30, 34 CrossRef.
- L. P. Kazansky and B. R. Mc Garvey, Coord. Chem. Rev., 1999, 188, 157 CrossRef CAS.
- N. Mizuno, K. Yamaguchi and K. Kamata, Coord. Chem. Rev., 2005, 249, 1944 CrossRef CAS.
- D. L. Long, E. Burkholder and L. Cronin, Chem. Soc. Rev., 2007, 36, 105 RSC.
- P. Gouzerh and A. Proust, Chem. Rev., 1998, 98, 77 CrossRef CAS PubMed.
- A. Proust, R. Thouvenot and P. Gouzerh, Chem. Commun., 2008, 1837 RSC.
- Y. G. Wei, B. B. Xu, C. L. Barnes and Z. H. Peng, J. Am. Chem. Soc., 2001, 123, 4083 CrossRef CAS PubMed.
- W. Guan, G. C. Yang, L. K. Yan and Z. M. Su, Inorg. Chem., 2006, 45, 7864 CrossRef CAS PubMed.
- G. C. Yang, W. Guan, L. K. Yan, Z. M. Su, L. Xu and E. B. Wang, J. Phys. Chem. B, 2006, 110, 23092 CrossRef CAS PubMed.
- I. Bar-Nahum, H. Cohen and R. Neumann, Inorg. Chem., 2003, 42, 3677 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frishch, J. Phys. Chem., 1994, 98, 11623 CrossRef CAS.
- T. Zhang, W. Guan, L. K. Yan, T. Y. Ma, J. Wang and Z. M. Su, Phys. Chem. Chem. Phys., 2015, 17, 5459 RSC.
- T. Zhang, W. Guan, S. Z. Wen, T. Y. Ma, L. K. Yan and Z. M. Su, J. Phys. Chem. C, 2014, 118, 29623 CAS.
- T. Zhang, N. N. Ma, L. K. Yan, T. Y. Ma and Z. M. Su, Dyes Pigm., 2014, 106, 105 CrossRef CAS.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999 CrossRef CAS PubMed.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270 CrossRef CAS.
- D. R. Kanis, M. A. Ratner and T. J. Marks, Chem. Rev., 1994, 94, 195 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215 CrossRef CAS.
- N. N. Ma, S. J. Li, L. K. Yan, Y. Q. Qiu and Z. M. Su, Dalton Trans., 2014, 43, 5069 RSC.
- T. Yanai, D. Tew and N. Handy, Chem. Phys. Lett., 2004, 393, 51 CrossRef CAS.
- H. Iikura, T. Tsuneda, T. Yanai and K. Hirao, J. Chem. Phys., 2001, 115, 3540 CrossRef CAS.
- N. N. Ma, C. G. Liu, Y. Q. Qiu, S. L. Sun and Z. M. Su, J. Comput. Chem., 2012, 33, 211 CrossRef CAS PubMed.
- T. Zhang, W. Guan, T. Y. Ma, L. Yan, L. K. Yan and Z. M. Su, Inorg. Chem. Front., 2015, 2, 544 RSC.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158 CrossRef CAS.
- D. Ravelli, D. Dondi, M. Fagnoni, A. Albini and A. Bagno, Phys. Chem. Chem. Phys., 2013, 15, 2890 RSC.
- D. Ravelli, D. Dondi, M. Fagnoni, A. Albini and A. Bagno, J. Comput. Chem., 2011, 32, 2983 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci and G. A. Petersson, et al., Gaussian 09W, revision A.02, Gaussian, Inc, Wallingford, CT, 2009 Search PubMed.
- D. D. Qi, L. J. Zhang, L. Wan, Y. X. Zhang, Y. Z. Bian and J. Z. Jiang, Phys. Chem. Chem. Phys., 2011, 13, 13277 RSC.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580 CrossRef CAS PubMed.
- J. L. Oudar and D. S. Chemla, J. Chem. Phys., 1977, 66, 2664 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra00146g |
| This journal is © The Royal Society of Chemistry 2016 |