
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Biological fluorination from the sea: discovery of a SAM-dependent nucleophilic fluorinating enzyme from the marine-derived bacterium Streptomyces xinghaiensis NRRL B24674†
Long
Ma
*abc,
Yufeng
Li
ab,
Lingpei
Meng
ab,
Hai
Deng
d,
Yuyin
Li
ab,
Qiang
Zhang
e and
Aipo
Diao
ab
aKey Laboratory of Industrial Fermentation Microbiology, Ministry of Education, School of Biotechnology, Tianjin University of Science & Technology, Tianjin 300457, China. E-mail: malong@tust.edu.cn; Fax: +86 22 60602298; Tel: +86 22 60602948
bTianjin Key Laboratory of Industry Microbiology, School of Biotechnology, Tianjin University of Science & Technology, Tianjin 300457, China. Tel: +86 133 446 7254
cBiomolecular Sciences Research Complex, EaStCHEM School of Chemistry, University of St Andrews, Fife KY16 9ST, UK. E-mail: lm228@st.andrews.ac.uk
dMarine Biodiscovery Centre, Department of Chemistry, University of Aberdeen, Meston Walk, Aberdeen AB24 3UE, UK
eTianjin 3rd Center Hospital, Tianjin 300170, China
First published on 2nd March 2016
Abstract
We have discovered and characterised a fluorinating enzyme from marine-derived Streptomyces xinghaiensis. It is able to install a fluorine atom into S-adenosyl-L-methionine (SAM) to form 5′-fluorodeoxy adenosine. To our knowledge, this is the first ever fluorinase unveiled from a marine source. It is ‘the most efficient’ fluorinase by far and, impressively, highly robust. Therefore it promises to be useful in different cases such as bio-transformation and synthetic biology.
Nature excites us by his hands to create a number of chemically complex structures with distinct bio-activities. It has been documented that over 70% of antibiotics entering clinical trials have been based on natural products.1 Marine organisms represent approximately half of the earth’s biodiversity. Up until now, amongst ∼5000 reported halogenated natural products,2 only six fluorinated products have been securely verified. This is very disproportionate to the abundance of fluorine in the earth, in despite of the fact that most fluorine elements are found in the earth crust and in insoluble forms. Importantly, fluorination of a molecule, especially a pharmaceutical, is of particular interest as halogenation is well known for its ability to tune the biophysical and bioactive properties to enhance drug efficacy through a range of mechanisms.3
In 2002, the first ever naturally occurring fluorinating enzyme (encoded by a flA gene) was isolated from Streptomyces cattleya, a terrestrial bacterium. It has been proven to have a unique ability to utilise inorganic fluorine ions and S-adenosyl-L-methionine (SAM) 1 to catalyse a SN2 biological nucleophilic reaction to generate 5′-fluorodeoxy adenosine (5′-FDA) 2 and L-methionine.4 It is the first committed enzyme in the fluorometabolites biosynthetic pathway Streptomyces cattleya. Another four enzymatic reactions are needed in turn to accomplish the biosynthesis of the final products of fluoroacetate (FAc) 6 and 4-fluorothreonine (4-FT) 7 in this organism. Fig. 1 describes this fluorometabolite biosynthetic pathway in detail. Taking one step further, in 2015, our work resulted in the discovery of a novel organofluorine natural product (2R3S4S)-5-fluoro-2,3,4-trihydroxypentanoic acid (5-FHPA) 10 from a Ghanaian Streptomyces isolate named Streptomyces sp. MA37. This also led to the elucidation of a new fdr gene cluster, encoding a branched pathway for biosynthesis of 5-FHPA 10 from a intermediate 5-fluoro-D-ribose (5-FDR) 8, as shown in Fig. 1.5 This disclosure deepens the understanding of de novo fluorinated product biosynthesis. 5-FHPA 10 is the first secure identification of a new fluorinated natural product since 1998.
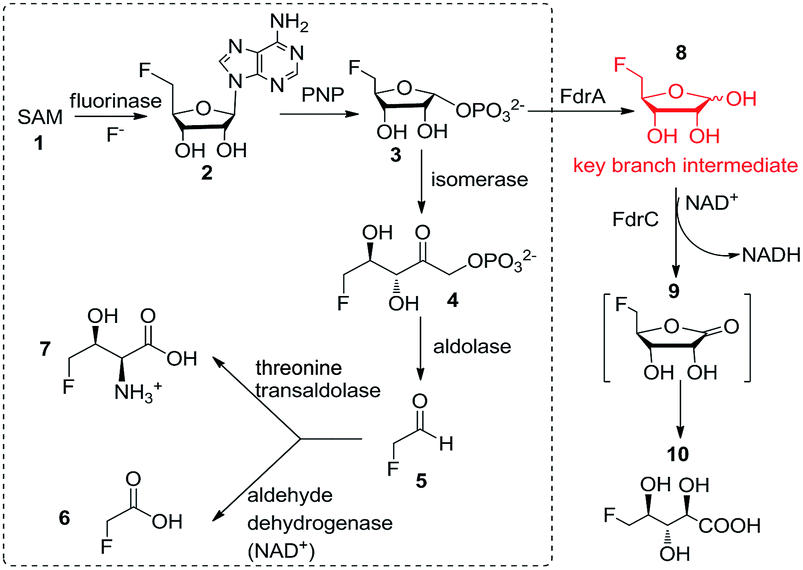 | ||
| Fig. 1 Known microbial biosynthetic pathways of fluorinated natural products so far. The one in the dotted line box denotes the biosynthetic pathway to FAc and 4-FT established in Streptomyces cattleya. The one outside the box depicts the branched fluorometabolite pathway in Streptomyces sp. MA37. | ||
The fluorinase from S. cattleya stands for a unique resource for fluorination in biology. This fluorinase gene remained a sole representative for roughly a decade. Nevertheless, in 2014 our work revealed three other putative fluorinase genes that appeared in the genomes database of sequenced microorganisms (Streptomyces sp. MA37, Actinoplanes sp. N902-109 and Norcardia brasiliensis). PCR amplification of these genes and in vitro tests have confirmed they are all functional fluorinases.6
The known fluorinases are all FAD (flavin adenine dinucleotide)-, heme-, vanadium- or iron-independent, unlike some other reported halogenases.7 They are reminiscent of two functionally related genes: the duf-62 family (∼30% in homology) and chlorinase (∼40% in homology). The above-mentioned three types of enzymes catalyse closely related reactions with SAM as substrates, as shown in Fig. S1.† The gene alignment, phylogenetic study of known fluorianses, duf-62 and chlorinase is shown in Fig. S2 and S3† respectively. A unique characteristic of fluorinases to chlorinase and duf-62 enzymes is a 22 amino acid loop, which is deemed as a specific diagnostic signature. Chlorinase is from the marine organism Salinospora tropica, which performs related transformations on SAM but with a nucleophile chlorine other than a fluoride ion.8 It is tasked to be the first enzyme in a biosynthetic pathway, leading to production of a potent anti-cancer agent salinosporamide A.9 There are over 200 kinds of duf-62 in available genomes, mostly found in extremophile organisms.10 They have been shown to catalyse a reaction between SAM and a hydroxide ion to generate adenosine using a different mechanism from fluorinase.11
Streptomyces xinghaiensis NRRL B-24674 was isolated in 2009 from a marine sediment sample at Xinghai Bay, Dalian, China. This strain produces a novel alkaloid named xinghaiamine A. It has been genome sequenced in 2011.12 Our previous work has shown that fermentation of Streptomyces xinghaiensis NRRL B24674 resulted in production of FAc 6 probed by 19F-NMR. This indicates an active fluorinase gene in vivo, which can also be implied by in silico gene annotation.13 In this work, we reported one putative fluorinase from this bacterial strain. We have deduced the open reading frame of it and found that it has an ∼84% identity to the original S. cattleya fluorinase. A recombinant gene vector for over-expression was constructed, followed by protein induction and purification (Fig. S4 and S5†). We performed a HPLC and LC-MS survey to prove that it is a functional fluorinase, as shown in Fig. 2A–C. A kinetic study of this S. xinghaiensis fluorinase has been carried out and fitted with Michaelis–Menten curves (Fig. 2D and E).
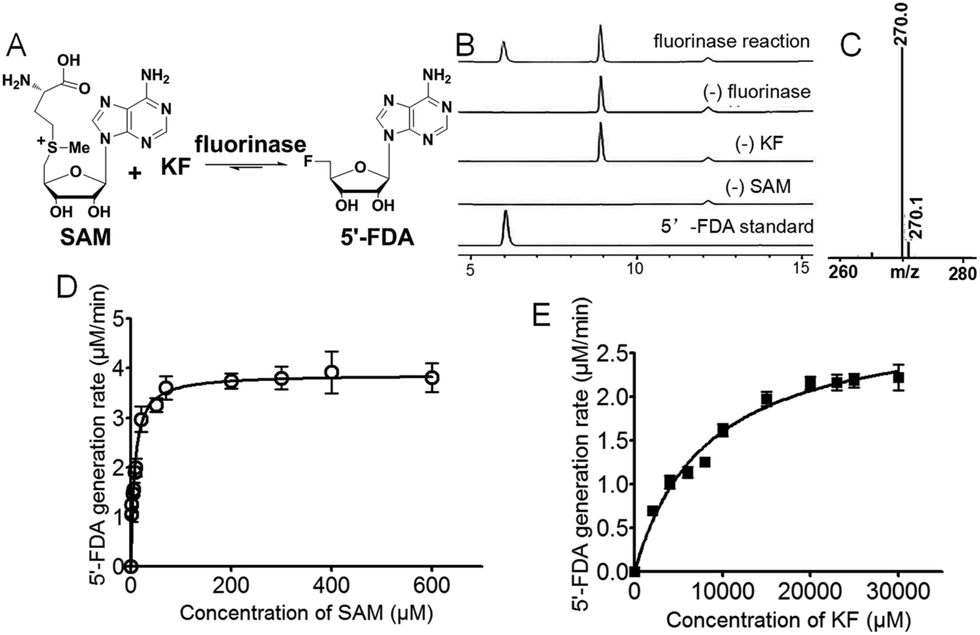 | ||
| Fig. 2 Identification of a fluorinase from Streptomyces xinghaiensis, which is able to catalyse the conversion of fluoride and SAM dual substrates to generate 5′-FDA. (A) The chemical reaction catalysed by fluorinase. (B) HPLC analysis of the reaction denoted in (A). (C) LC-MS profile of the enzymatic reaction product 5′-FDA. The observed value (m/z [M + H]+ = 270.0) is consistent with the calculated value (m/z [M + H]+ = 270.10). Error bars are standard deviations for experiments performed in triplicate. | ||
In order to do so, we established a standard curve for 5′-FDA quantification based on HPLC measurements (Fig. S6†). The Km with regard to SAM is 7.04 ± 0.94 μM; that to KF is rather large, reaching 8.20 ± 0.96 mM. It is highly agreeable with previously reported fluorinases, for which are all ‘slow’ and the Km for KF is at the millimolar level.14 The Vmax is 3.88 ± 0.11 μM min−1, in terms of SAM. The turnover number (kcat) was 0.277 ± 0.007 min−1, and the specificity constant is equal to 39.5 ± 1.51 mM−1 min−1. We also performed a side-by-side comparison of S. xinghaiensis fluorinase with all other known fluorinases which is shown in Table 1 and Fig. S10.† From it we can see that the original S. cattleya enzyme is the least efficient, while the S. xinghaiensis fluorinase is the most efficient one, as evidenced by the fact that its specificity constant is around one order of magnitude larger than the rest. It has been previously reported that in the presence of L-amino acid oxidase (L-AAO), S. cattleya fluorinase is capable of utilising halogen ions Cl− to form 5′-chloro-5′-deoxyadenosine (5′-CIDA).15 We have observed that S. xinghaiensis fluorinase is able to perform a reaction of the same kind. But this must require the presence of L-AAO to shift the equilibrium of the reaction towards the organochlorine product (Fig. 3).
| Fluorinase source | V max (μM min−1) | SAM Km (μM) | k cat = Vmax/C (min−1) | Specificity constant = kcat/Km (mM−1 min−1) |
|---|---|---|---|---|
| Streptomyces xinghaiensis | 3.88 ± 0.11 | 7.04 ± 0.94 | 0.277 ± 0.007 | 39.5 ± 1.51 |
| Streptomyces cattleya | 1.17 ± 0.06 | 29.4 ± 5.80 | 0.084 ± 0.005 | 2.84 ± 0.14 |
| Streptomyces sp. MA37 | 3.63 ± 0.13 | 86.0 ± 11.3 | 0.260 ± 0.004 | 3.02 ± 0.19 |
| Nocardia brasiliensis | 1.77 ± 0.07 | 30.4 ± 4.24 | 0.128 ± 0.004 | 4.22 ± 0.42 |
| Actinoplanes sp. N902-109 | 2.78 ± 0.15 | 43.1 ± 7.99 | 0.197 ± 0.003 | 4.58 ± 0.50 |
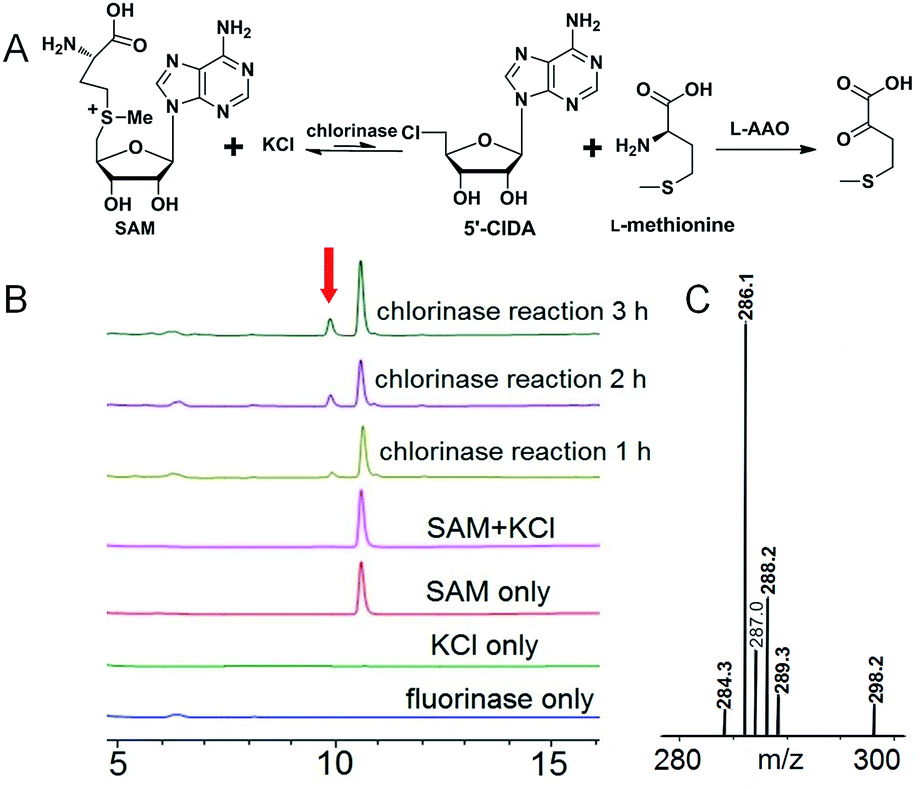 | ||
| Fig. 3 Fluorinase from Streptomyces xinghaiensis NRRL B24674 is also a chlorinase, which is able to catalyse conversion of chloride and SAM to 5′-CIDA and L-methionine in the presence of L-amino acid oxidase. (A) The chemical reaction catalysed by fluorinase. (B) HPLC analysis of the reaction denoted in (A), the red arrow indicates 5′-CIDA production. (C) LC-MS profile of the enzymatic reaction product 5′-CIDA. The observed values (m/z [M + H]+ 35Cl = 286.1, 37Cl = 288.2) are consistent with the calculated values (m/z [M + H]+ 35Cl = 286.07, 37Cl = 288.07). | ||
Next, we conducted a mutational study to find out the catalytically important amino acid residues (primers used are listed in Table S1†). Error bars are standard deviations for experiments performed in triplicate.
Fig. 4 shows mutational studies in a modeled S. xinghaiensis fluorinase structure. All the mutants showed some distortions in terms of secondary structure in comparison to the wild type enzyme; however, not to a great extent. This can be empirically reflected by far-UV circular dichroism (CD) spectroscopy recorded in the range of 190 to 260 nm (Fig. S7†). It has been proposed that S158 is a key residue for hydrogen bonding with fluorine ions. This can be mirrored by the current experiment, in which S158A only retains less than 20% activity compared with the WT one. D16 is crucial for substrate SAM binding and the F156 residue provides the rear of the hydrophobic pocket which is necessary to assist desolvation of fluoride ions. D16E and 156A mutants are detrimental to enzyme activity. T80A and Y77A severely abolish activity in our case, consistent with the mechanism that T80 and Y77 are able to facilitate halide binding. The mutated amino acids and relevant positions are shown in Fig. S8.†
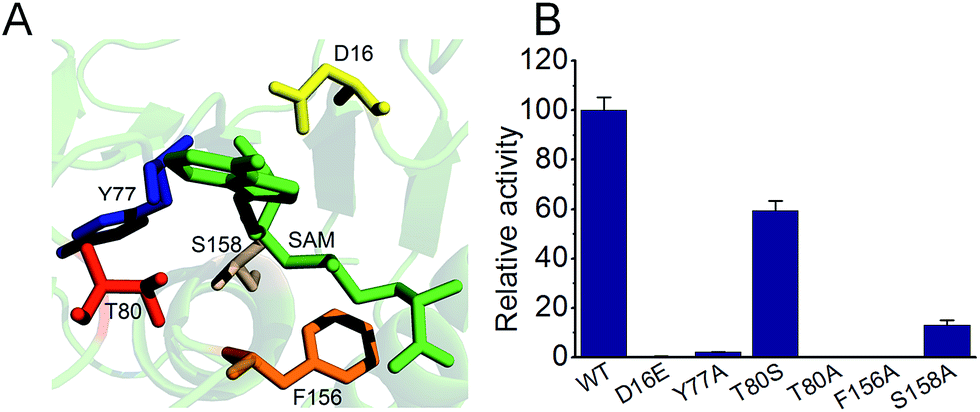 | ||
| Fig. 4 Mutational studies of S. xinghaiensis fluorinase. (A) Mutation locations together with SAM. (B) The relative activities of all mutants. | ||
For this ‘most efficient ever’ S. xinghaiensis fluorianse, we would like to evaluate its stability and optimise its reaction conditions. Stability is an important property for all enzymes, particularly for those encountered in industrial processes. Optimal buffer ion and pH conditions always lead to great benefits including prolonging catalyst lifetime and increasing enzyme tolerance to external stress.16 Thermal shift analysis (TFA) is a quick and easy-to-use method for probing protein stability (mostly influenced by buffer compositions, metal ions, and pH, etc.), solubility and protein–ligand interactions. It can be best performed using a conventional real-time PCR. Simply, when a protein undergoes thermal denaturation, its hydrophobic regions can bind the environmentally sensitive dye SYPRO Orange, which produces enhanced fluorescence. The fluorescence intensity correlates well with protein melting.17 We did observe that ligands such as adenosine and the enzyme reaction product 5′-FDA enhance the protein stability and the wild-type enzyme is more stable than all mutants (Fig. S9 and Table S2†). We also tested different buffers and found Tris–HCl buffer (pH 7.0) is better for maintaining stability (Table S3†). We conducted a series of experiments to examine the feasibility of S. xinghaiensis fluorianse as a biocatalyst to catalyse F–C bond formation. As shown in Fig. 5A, the optimal temperature for S. xinghaiensis fluorianse is around 40 °C as expected. This is also a common temperature set for most enzymatic bio-transformation reactions. The pH value seems crucial for its activity. Intriguingly, this enzyme was rather active in an acidic environment with a pH value as low as 4. With a basic condition (pH 8–9.5), its activity drops significantly (Fig. 5B). This observation renders the possibility of this enzyme to fulfil a bio-transformation task in aqueous buffer with pH within the range from 4 to 8. Fig. 5C shows that fluorination activity is highest in the ‘control’ condition (without EDTA and metal ions), and the addition of EDTA (to chelate metal ions) makes this enzyme considerably less active. This indicates that this fluorinase is possibly metal ion dependent. Cu2+ and Zn2+ can considerably inhibit activity. Furthermore, to be a candidate for industrial purposes, the enzyme should possess longevity in the buffer and be stable at ambient temperature. To test this, S. xinghaiensis fluorinase was placed at 25 °C in a 50 mM Tris–HCl buffer (pH 7.0). The enzyme activity was examined at different time points. Impressively, the enzyme showed excellent stability over 4 days, in which ∼87% of the activity remains in the 4th day (Fig. 5D). Last, for a industrially relevant biocatalyst, a good expression platform is required. In the current study, we used E. coli to over-express this enzyme and have found that it is manageable to obtain ∼100 mg of purified protein per gram of dried E. coli cells, which can be acquired from a 2 liter cell culture. Of course, other practical expression systems could be further developed for its production.
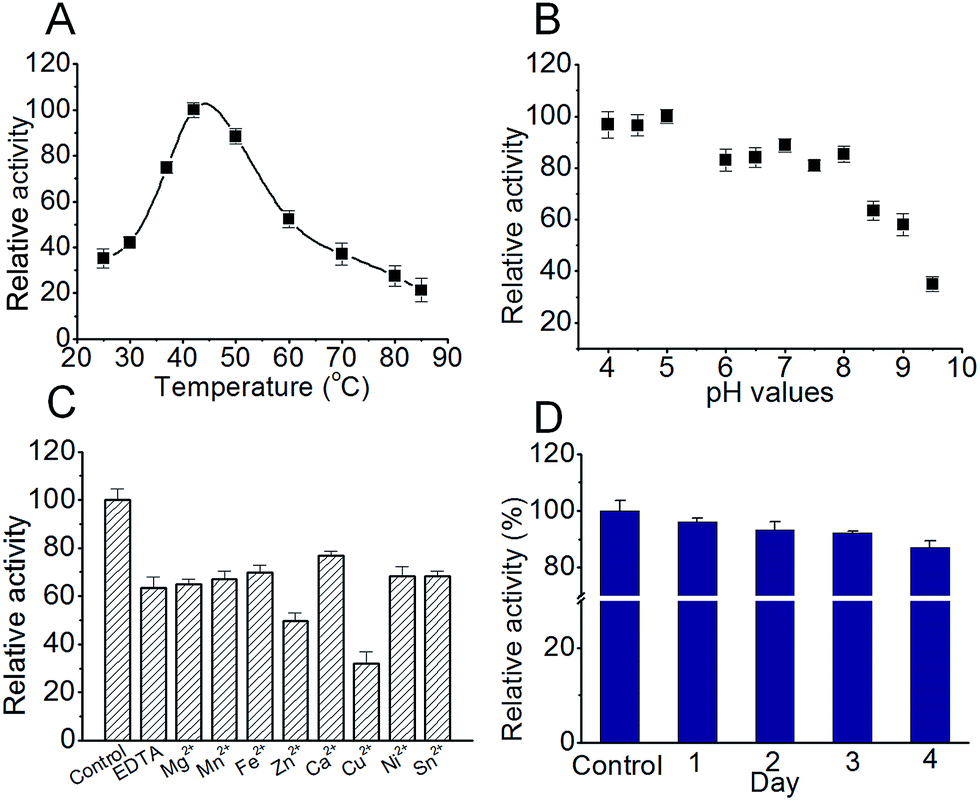 | ||
| Fig. 5 Buffer composition, optimal pH, temperature and ion condition for a fluorinating reaction catalysed by Streptomyces xinghaiensis fluorinase. Error bars are standard deviations for experiments performed in triplicate. | ||
Naturally-derived products (small molecules, proteins and nucleic acids) have been recognised to be of high importance in sensing, diagnosis and treatment.18 Our report of this marine-originated fluorinase, to our knowledge, is the first fluorinating enzyme discovered from a marine organism, which would expand the repertoire of exotic biological halogenation. It is also ‘the most efficient’ one. This might be because soluble fluoride ions are more prevalent in the sea than terrestrial sources, thus accelerating the evolution of fluorine biochemistry. We have also presented some fundamental data, which enable this enzyme to be more amenable for practical uses. In addition, theoretically and practically, halogenase, as biocatalysts can be used for generating halogenated natural product analogs with improved properties, which is highly industrially and bio-technologically relevant.19 In this work, we have afforded a most efficient fluorinase, which could be engineered or used as it is to fulfil bio-transformation purposes (e.g. fluorinated nucleic acid derivatives). The fluorinase gene can be used as a component in synthetic biology for generating the ‘fluorinated version’ of bio-active compounds using a microbial factory.20 Surely, further studies of this enzyme (gene) will be explored both experimentally and computationally.21
Acknowledgements
We thank Prof. David O’Hagan and Dr Qingzhi Zhang (University of St Andrews, UK) for their helpful discussion and for providing the synthetic 5′-FDA sample. This work is supported by National Natural Science Foundation of China (No. 81503086), a starting funding (No. 20140520) from Tianjin University of Science & Technology, a research funding of “1000 Talents Plan” of Tianjin (to LM) and Foundation of Key Laboratory of Industrial Fermentation Microbiology of Ministry of Education and Tianjin Key Lab of Industrial Microbiology (No. 2015IM106).Notes and references
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2012, 75, 311–335 CrossRef CAS PubMed.
- G. Gribble, Mar. Drugs, 2015, 13, 4044–4136 CrossRef CAS PubMed.
- (a) K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881–1886 CrossRef PubMed; (b) C. D. Murphy and G. Sandford, Expert Opin. Drug Metab. Toxicol., 2015, 11, 589–599 CrossRef CAS PubMed; (c) Y. Lu, Y. Liu, Z. Xu, H. Li, H. Liu and W. Zhu, Expert Opin. Drug Discovery, 2012, 7, 375–383 CrossRef CAS PubMed; (d) C. Odar, M. Winkler and B. Wiltschi, Biotechnol. J., 2015, 10, 427–446 CrossRef CAS PubMed; (e) L. Ma and J. Liu, J. Ethnopharmacol., 2014, 158, 358–363 CrossRef CAS PubMed.
- (a) D. O’Hagan, C. Schaffrath, S. L. Cobb, J. T. G. Hamilton and C. D. Murphy, Nature, 2002, 416, 279 CrossRef PubMed; (b) C. Dong, F. Huang, H. Deng, C. Schaffrath, J. B. Spencer, D. O’Hagan and J. H. Naismith, Nature, 2004, 427, 561–565 CrossRef CAS PubMed.
- (a) L. Ma, A. Bartholome, M. H. Tong, Z. Qin, Y. Yu, T. Shepherd, K. Kyeremeh, H. Deng and D. O’Hagan, Chem. Sci., 2015, 6, 1414–1419 RSC; (b) Y. Wang, Z. Deng and X. Qu, F1000Research, 2014, 61, 1 CAS.
- H. Deng, L. Ma, N. Bandaranayaka, Z. Qin, G. Mann, K. Kyeremeh, Y. Yu, T. Shepherd, J. H. Naismith and D. O’Hagan, ChemBioChem, 2014, 15, 364–368 CrossRef CAS PubMed.
- (a) J. L. R. Anderson and S. K. Chapman, Mol. BioSyst., 2006, 2, 350–357 RSC; (b) A. Butler and M. Sandy, Nature, 2009, 460, 848–854 CrossRef CAS PubMed; (c) H. M. Senn, Front. Chem., 2014, 2, 98 Search PubMed.
- A. S. Eustaquio, F. Pojer, J. P. Noel and B. S. Moore, Nat. Chem. Biol., 2008, 4, 69–74 CrossRef CAS PubMed.
- L. Ma and A. Diao, Adv. Anticancer Agents Med. Chem., 2015, 15, 298–306 CrossRef CAS.
- H. Deng, C. H. Botting, J. T. G. Hamilton, R. J. M. Russell and D. O’Hagan, Angew. Chem., Int. Ed., 2008, 47, 5357–5361 CrossRef CAS PubMed.
- H. Deng and D. O’Hagan, Curr. Opin. Chem. Biol., 2008, 12, 582–592 CrossRef CAS PubMed.
- (a) X. Q. Zhao, W. J. Li, W. C. Jiao, Y. Li, W. J. Yuan, Y. Q. Zhang, H. P. Klenk, J. W. Suh and F. W. Bai, Int. J. Syst. Evol. Microbiol., 2009, 59, 2870–2874 CrossRef CAS PubMed; (b) X. Zhao and T. Yang, J. Bacteriol., 2011, 193, 5543 CrossRef CAS PubMed; (c) W. Jiao, F. Zhang, X. Zhao, J. Hu and J.-W. Suh, PLoS One, 2013, 8, e75994 CAS.
- S. Huang, L. Ma, M. H. Tong, Y. Yu, D. O’Hagan and H. Deng, Org. Biomol. Chem., 2014, 12, 4828–4831 CAS.
- X. Zhu, D. A. Robinson, A. R. McEwan, D. O’Hagan and J. H. Naismith, J. Am. Chem. Soc., 2007, 129, 14597–14604 CrossRef CAS PubMed.
- H. Deng, S. L. Cobb, A. R. McEwan, R. P. McGlinchey, J. H. Naismith, D. O’Hagan, D. A. Robinson and J. B. Spencer, Angew. Chem., Int. Ed., 2006, 45, 759–762 CrossRef CAS PubMed.
- (a) C. B. Poor, M. C. Andorfer and J. C. Lewis, ChemBioChem, 2014, 15, 1286–1289 CrossRef CAS PubMed; (b) L. D. Finger, N. Patel, A. Beddows, L. Ma, J. C. Exell, E. Jardine, A. C. Jones and J. A. Grasby, Nucleic Acids Res., 2013, 41, 9839–9847 CrossRef PubMed; (c) L. Ma, X. Wu, G. G. Wilson, A. C. Jones and D. T. F. Dryden, Biochem. Biophys. Res. Commun., 2014, 449, 120–125 CrossRef CAS PubMed.
- (a) F. H. Niesen, H. Berglund and M. Vedadi, Nat. Protoc., 2007, 2, 2212–2221 CrossRef CAS PubMed; (b) K. Huynh and C. L. Partch, in Curr. Protoc. Protein Sci., John Wiley & Sons, Inc., 2001, 79:28.9.1-28.9.14 Search PubMed.
- (a) L. Ma and S. L. Cockroft, ChemBioChem, 2010, 11, 25–34 CrossRef CAS PubMed; (b) C. P. Wilson, C. Boglio, L. Ma, S. L. Cockroft and S. J. Webb, Chem.–Eur. J., 2011, 17, 3465–3473 CrossRef CAS PubMed; (c) L. Ma, K. Chen, D. J. Clarke, C. P. Nortcliffe, G. G. Wilson, J. M. Edwardson, A. J. Morton, A. C. Jones and D. T. F. Dryden, Nucleic Acids Res., 2013, 41, 4999–5009 CrossRef CAS PubMed; (d) L. Ma and A. Diao, Chem. Commun., 2015, 51, 10233–10235 RSC; (e) L. Ma, G. Wu, Y. Li, P. Qin, L. Meng, H. Liu, Y. Li and A. Diao, Nanoscale, 2015, 7, 18044–18048 RSC; (f) T. Sabir, A. Toulmin, L. Ma, A. C. Jones, P. McGlynn, G. F. Schroder and S. W. Magennis, J. Am. Chem. Soc., 2012, 134, 6280–6285 CrossRef CAS PubMed.
- (a) D. R. M. Smith, S. Gruschow and R. J. M. Goss, Curr. Opin. Chem. Biol., 2013, 17, 276–283 CrossRef CAS PubMed; (b) M. C. Walker and M. C. Y. Chang, Chem. Soc. Rev., 2014, 43, 6527–6536 RSC; (c) S. A. Shepherd, C. Karthikeyan, J. Latham, A.-W. Struck, M. L. Thompson, B. R. K. Menon, M. Q. Styles, D. Leys and J. Micklefield, Chem. Sci., 2015, 6, 3454–3460 RSC; (d) S. Brown and S. E. O’Connor, ChemBioChem, 2015 Search PubMed; (e) J. Fernández-Lucas, Appl. Microbiol. Biotechnol., 2015, 99, 4615–4627 CrossRef PubMed.
- (a) A. S. Eusthquio, D. O’Hagan and B. S. Moore, J. Nat. Prod., 2010, 73, 378–382 CrossRef PubMed; (b) M. C. Walker and M. C. Y. Chang, Chem. Soc. Rev., 2014, 43, 6527–6536 RSC.
- (a) L. C. Blasiak and C. L. Drennan, Acc. Chem. Res., 2009, 42, 147–155 CrossRef CAS PubMed; (b) A. Timmins and S. P. de Visser, Advances in Protein Chemistry and Structural Biology, Academic Press, 2015, vol. 100, pp. 113–151 Search PubMed; (c) H. M. Senn, D. O’Hagan and W. Thiel, J. Am. Chem. Soc., 2005, 127, 13643–13655 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra00100a |
| This journal is © The Royal Society of Chemistry 2016 |