
Efficient and selective glucosylation of prenylated phenolic compounds by Mucor hiemalis†
Shuai Ji‡
,
Wen-Fei Liang‡,
Zi-Wei Li,
Jin Feng,
Qi Wang,
Xue Qiao and
Min Ye*
State Key Laboratory of Natural and Biomimetic Drugs, School of Pharmaceutical Sciences, Peking University, 38 Xueyuan Road, Beijing 100191, China. E-mail: yemin@bjmu.edu.cn; Fax: +86 10 82802024; Tel: +86 10 82801516
First published on 15th February 2016
Abstract
A highly regio- and stereo-specific biocatalytic reaction to produce the glucosides of natural prenylated phenolic compounds by Mucor hiemalis CGMCC 3.14114 is described. M. hiemalis could efficiently catalyze β-O-glucosylation of the isoprenyl-neighboring hydroxyl group, rather than other hydroxyls with low steric hindrance. This reaction was demonstrated by 28 compounds with different basic skeletons. The conversion rate was above 90% for most compounds. Scaled-up biotransformations of 15 substrates yielded 17 products, including 12 new compounds. These products showed enhanced water solubility and improved intestinal absorption in a Caco-2 cell monolayer model. This reaction provides a facile and efficient approach to synthesize the glucosides of prenylated phenolics, which are rare in natural products.
Introduction
Prenylated phenolic compounds usually show significant biological activities, including antitumor, anti-inflammatory, antioxidant, antibacterial, antiviral, and estrogenic activities.1,2 They are widely present as secondary metabolites of higher plants, particularly those derived from Leguminosae and Moracece families.3 Recently, we have isolated a series of prenylated flavonoids and coumarins from Glycyrrhiza uralensis Fisch., which is used as the popular Chinese herbal medicine Gan-Cao.4–6 Some of these compounds show potent bioactivities. For instance, glycycoumarin (1) possesses significant hepatoprotective, anti-hepatitis C virus, and antibacterial activities.7–9 However, medicinal use of these lipophilic compounds is usually hindered by poor water solubility and low bioavailability.Glycosylation is an effective approach to increase water solubility of natural products, as well as to improve their pharmacological properties.10 Although a number of chemical glycosylation tactics have been established, the glycosylation of vulnerable phenolic compounds with multiple functional groups is still a big challenge.11 Particularly, the reaction yield and selectivity are generally far from ideal.12 Thus far, few reports are available on the glycosylation of prenylated phenolic compounds, possibly due to steric hindrance of the isoprenyl group. Biocatalysis provides an alternative approach for structural modification of natural products. A number of microbial strains have been reported to catalyze stereo- and regio-selective glycosylation reactions under mild conditions.13
In this work, we report highly efficient and selective glucosylation of prenylated phenolic compounds by the filamentous fungus Mucor hiemalis CGMCC 3.14114. M. hiemalis could specifically catalyze β-O-glucosylation of ortho-OH of the isoprenyl group with conversion rates of above 90% to obtain a series of new compounds. Moreover, water solubility, bioavailability, and bioactivities of the biotransformed products were tested.
Experimental section
General information
The NMR spectra were recorded at 400 MHz for 1H and 100 MHz for 13C on a Bruker AVANCE III-400 spectrometer in DMSO-d6 using TMS as the reference, except for 1a and 10a (600 MHz for 1H and 150 MHz for 13C). HRESIMS spectra were obtained on a Bruker APEX IV FT-MS spectrometer. Oasis HLB cartridges (20 cm3, 1 g) were purchased from Waters (MA, USA). Semi-preparative HPLC was performed on an Agilent 1200 instrument equipped with a Zorbax SB-C18 column (9.4 × 250 mm, 5 μm, Agilent, USA).HPLC-DAD-ESI-MSn analysis was performed on an Agilent 1100 instrument coupled with a Finnigan LCQ advantage ion trap mass spectrometer (Thermo Fisher Scientific, Waltham, MA). Samples were separated on an Eclipse XDB-C18 column (4.6 × 250 mm, 5 μm, Agilent, USA) protected with a Zorbax Extend-C18 guard column (4.6 × 12.5 mm, 5 μm). The column temperature was 30 °C. The mobile phase consisted of acetonitrile (A) and water containing 0.1% (v/v) formic acid (B). A linear gradient elution program was used: 0 min, 19% A; 8 min, 28% A; 16 min, 28% A; 17 min, 35% A; 21 min, 50% A; 26 min, 50% A; 38 min, 75% A; 48 min, 95% A; 53 min, 95% A. The flow rate was 1.0 mL min−1. UV spectra were obtained by scanning from 200 to 400 nm. For MS analysis, the effluent was introduced into the ESI source of mass spectrometer at 0.2 mL min−1 via a T-union splitter. The mass spectrometer was operated in the (−)-ESI mode.
Chemicals and reagents
Among the substrates used in this study, 24 compounds (1, 2, 4–16, 18, 19, 21, 23, 29, 31–34) were isolated from licorice by the authors. Compounds 3, 22, 24–28, 30 and 35 were from our compound library. Compound 20 was purified by acid hydrolysis of icariin. Compound 17 was a dehydrogenated product of licoricidin (16) catalyzed by DDQ (dichloro-5,6-dicyano-1,4-benzoquinone, Sigma). The purities were above 98% according to HPLC-UV analysis.Analytical grade reagents were purchased from Beijing Chemical Corporation (Beijing, China). For HPLC and LC/MS analysis, HPLC grade methanol, acetonitrile and formic acid (Mallinkrodt, Phillipsburg, NJ, USA) were used. High-purity nitrogen (99.9%) and helium (99.99%) were from Gas Supple Center of Peking University Health Science Center (Beijing, China). Standard samples of D-glucose, D-galactose, and β-glucosidase were from Sigma-Aldrich (Shanghai, China).
Biotransformation process and products purification
The fungal strains were obtained from China General Microbiological Culture Collection Center (CGMCC, Beijing, China), and were maintained on agar slope at 4 °C. For analytical-scale biotransformations, 250 mL flasks containing 100 mL of potato culture medium were used. After the substrates (1 mg) were added and incubated for three days, the culture medium was extracted and analyzed by HPLC and LC-MS. The preparative-scale biotransformations were carried out in 1000 mL Erlenmeyer flasks with 400 mL of potato culture medium. Mucor hiemalis CGMCC 3.14114 was incubated in the flasks at room temperature on a rotary shaker for three days to reach the exponential phase. Then the substrates in DMSO were added into the culture media. After three additional days of incubation, the supernatant was extracted with an equal volume of ethyl acetate (EtOAc) for three times. The organic phase was combined and evaporated to dryness. The pigments in the resulted extracts were removed by solid phase extraction (Oasis HLB, 20 cm3, 1 g) eluted with MeOH–H2O (H2O, 20 mL; 60% MeOH, 20 mL; MeOH, 20 mL), or silica gel column chromatography eluted with CHCl3–MeOH (0–50%, v/v). Then the fractions were purified by semi-preparative HPLC to obtain the products. The known compounds were identified as wighteone 7-O-β-D-glucoside (5a),14 luteone 7-O-β-D-glucoside (6a),15 licoflavonol 7-O-β-D-glucoside (18a),16 icariside (20a),17 corylifolinin 4′-O-β-D-glucoside (26a),18 astragalin (29a)19 and isoquercitrin (30a)20 by comparing their spectral data with those of the literatures.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ε) 335 (3.56) nm; IR (KBr) νmax 3411, 2917, 1609, 1097, 1076 cm−1; HRESIMS m/z 531.1856 [M + H]+ (calcd for C27H31O11, 531.1861). 1H NMR (600 MHz, DMSO-d6) and 13C NMR (150 MHz, DMSO-d6), see Table 1.
ε) 335 (3.56) nm; IR (KBr) νmax 3411, 2917, 1609, 1097, 1076 cm−1; HRESIMS m/z 531.1856 [M + H]+ (calcd for C27H31O11, 531.1861). 1H NMR (600 MHz, DMSO-d6) and 13C NMR (150 MHz, DMSO-d6), see Table 1.
| No. | 1aa | 7a | 9a | 10aa | ||||
|---|---|---|---|---|---|---|---|---|
| δH (J in Hz) | δC, type | δH (J in Hz) | δC, type | δH (J in Hz) | δC, type | δH (J in Hz) | δC, type | |
| a Data were recorded at 600 MHz for 1H NMR and 150 MHz for 13C NMR. | ||||||||
| 2 | 159.9, C | 8.48, s | 154.7, CH | 8.16, s | 155.2, CH | 8.38, s | 154.5, CH | |
| 3 | 122.0, C | 122.0, C | 119.6, C | 121.9, C | ||||
| 4 | 7.87, s | 135.9, CH | 180.9, C | 179.9, C | 180.0, C | |||
| 5 | 154.7, C | 159.8, C | 161.9, C | 162.0, C | ||||
| 6 | 120.1, C | 6.62, s | 98.2, CH | 6.23, d (1.8) | 99.0, CH | 6.23, d (2.0) | 99.1, CH | |
| 7 | 158.3, C | 160.5, C | 164.3, C | 164.4, C | ||||
| 8 | 6.95, s | 98.4, CH | 108.2, C | 6.40, d (1.8) | 93.8, CH | 6.40, d (2.0) | 93.7, CH | |
| 9 | 152.8, C | 154.1, C | 157.6, C | 157.5, C | ||||
| 10 | 108.1, C | 105.9, C | 104.3, C | 104.4, C | ||||
| 1′ | 113.2, C | 121.1, C | 107.1, C | 122.6, C | ||||
| 2′ | 156.1, C | 7.40, d (8.4) | 130.2, CH | 151.1, C | 7.01 d (0.8) | 121.8, CH | ||
| 3′ | 6.38, d (2.4) | 102.7, CH | 6.82, d (8.4) | 115.1, CH | 112.6, C | 145.2, C | ||
| 4′ | 158.6, C | 157.5, C | 153.1, C | 142.0, C | ||||
| 5′ | 6.28, dd (8.4, 2.4) | 106.3, CH | 6.82, d (8.4) | 115.1, CH | 6.72, d (8.4) | 111.1, CH | 120.6, C | |
| 6′ | 7.14, d (8.4) | 131.6, CH | 7.40, d (8.4) | 130.2, CH | 7.05, d (8.4) | 131.3, CH | 7.15 d (0.8) | 121.7, CH |
| 1′′ | 3.46, m; 3.27, m | 22.5, CH2 | 3.58, m; 3.30, br s | 21.3, CH2 | 6.79, d (9.6) | 116.8, CH | 6.42, d (10.0) | 117.9, CH |
| 2′′ | 5.21, t (7.2) | 122.6, CH | 5.17, br s | 122.2, CH | 5.70, d (9.6) | 129.6, CH | 5.79, d (10.0) | 131.5, CH |
| 3′′ | 130.8, C | 131.2, C | 76.0, C | 76.4, C | ||||
| 4′′ | 1.63, s | 25.5, CH3 | 1.62, s | 25.5, CH3 | 1.30, s | 27.6, CH3 | 1.41, s | 27.6, CH3 |
| 5′′ | 1.75, s | 17.8, CH3 | 1.78, s | 17.8, CH3 | 1.30, s | 27.6, CH3 | 1.40, s | 27.3, CH3 |
| 1′′′ | 4.98, d (7.8) | 100.7, CH | 5.00, d (6.4) | 100.5, CH | 4.91, d (7.2) | 100.9, CH | 4.83, d (7.2) | 100.7, CH |
| 2′′′ | 3.31, d (6.0) | 73.4, CH | 3.30, br s | 73.4, CH | 3.25, m | 73.3, CH | 3.28, d (5.6) | 73.2, CH |
| 3′′′ | 3.31, d (6.0) | 76.8, CH | 3.30, br s | 76.6, CH | 3.25, m | 76.9, CH | 3.28, d (5.6) | 76.5, CH |
| 4′′′ | 3.18, br s | 69.8, CH | 3.16, d (4.8) | 69.7, CH | 3.16, br s | 69.8, CH | 3.18, br s | 69.7, CH |
| 5′′′ | 3.46, m | 77.2, CH | 3.41, m | 77.2, CH | 3.27, m | 77.1, CH | 3.33, m | 77.1, CH |
| 6′′′ | 3.75, d (10.2); 3.47, m | 60.8, CH2 | 3.71, m; 3.44, m | 60.6, CH2 | 3.68, d (10.8); 3.45, m | 60.7, CH2 | 3.69, d (11.6); 3.50, m | 60.7, CH2 |
| OCH3 | 3.79, s | 63.0, CH3 | ||||||
ε) 284 (3.63) nm; IR (KBr) νmax 3405, 2921, 1603, 1113, 1045, 943, 598 cm−1; HRESIMS m/z 513.1760 [M + H]+ (calcd for C27H29O10, 513.1755). 1H NMR (400 MHz, DMSO-d6) and 13C NMR (100 MHz, DMSO-d6), see Table 2.
| No. | 4a | 19a | 22a | 24a | ||||
|---|---|---|---|---|---|---|---|---|
| δH (J in Hz) | δC, type | δH (J in Hz) | δC, type | δH (J in Hz) | δC, type | δH (J in Hz) | δC, type | |
| 2 | 5.46, s | 64.2, CH2 | 147.4, C | 155.4, C | 5.45, m | 79.2, CH | ||
| 3 | 107.4, C | 136.0, C | 133.3, C | 3.16, m; 2.64, br d (16.0) | 43.2, CH2 | |||
| 4 | 144.2, C | 176.1, C | 177.8, C | 190.6, C | ||||
| 5 | 152.9, C | 159.4, C | 158.5, C | 7.48, s | 126.1, CH | |||
| 6 | 117.6, C | 111.8, C | 6.30, s | 98.3, CH | 124.1, C | |||
| 7 | 156.3, C | 160.6, C | 161.7, C | 161.4, C | ||||
| 8 | 6.57, s | 100.1, CH | 6.87, s | 93.1, CH | 107.0, C | 6.68, s | 102.4, CH | |
| 9 | 152.7, C | 154.0, C | 153.6, C | 161.2, C | ||||
| 10 | 104.9, C | 104.4, C | 104.0, C | 114.6, C | ||||
| 1′ | 116.9, C | 122.2, C | 122.8, C | 129.1, C | ||||
| 2′ | 155.5, C | 8.07, d (8.8) | 129.6, CH | 8.23, d (9.2) | 130.7, CH | 7.33, d (8.4) | 128.4, CH | |
| 3′ | 7.00, d (2.0) | 98.0, CH | 6.94, d (8.8) | 115.5, CH | 7.06, d (9.2) | 113.7, CH | 6.79, d (8.4) | 115.1, CH |
| 4′ | 156.1, C | 156.8, C | 161.2, C | 157.7, C | ||||
| 5′ | 6.77, dd; (8.4, 2.0) | 112.4, CH | 6.94, d (8.8) | 115.5, CH | 7.06, d (9.2) | 113.7, CH | 6.79, d (8.4) | 115.1, CH |
| 6′ | 7.34, d (8.4) | 119.2, CH | 8.07, d (8.8) | 129.6, CH | 8.23, d (9.2) | 130.7, CH | 7.33, d (8.4) | 128.4, CH |
| 1′′ | 3.44, m; 3.25, m | 22.2, CH2 | 3.44, m; 3.20, m | 21.2, CH2 | 2.75, t (8.4) | 17.4, CH2 | 3.27, m | 27.2, CH2 |
| 2′′ | 5.21, t (6.4) | 123.5, CH | 5.23, t (7.2) | 121.6, CH | 1.56, t (8.4) | 42.9, CH2 | 5.32, m | 122.2, CH |
| 3′′ | 129.9, C | 130.7, C | 68.8, C | 132.1, C | ||||
| 4′′ | 1.63, s | 25.5, CH3 | 1.62, s | 25.5, CH3 | 1.17, s | 29.1, CH3 | 1.67, s | 25.6, CH3 |
| 5′′ | 1.75, s | 17.7, CH3 | 1.75, s | 17.8, CH3 | 1.17, s | 29.1, CH3 | 1.71, s | 17.6, CH3 |
| 1′′′ | 4.80, d (7.2) | 101.0, CH | 5.04, d (6.8) | 100.4, CH | 5.50, d (7.2) | 101.0, CH | 4.95, m | 100.1, CH |
| 2′′′ | 3.28, m | 73.4, CH | 3.31, m | 73.4, CH | 3.22, m | 74.2, CH | 3.27, m | 73.3, CH |
| 3′′′ | 3.27, m | 76.7, CH | 3.30, m | 76.8, CH | 3.22, m | 76.4, CH | 3.27, m | 76.5, CH |
| 4′′′ | 3.14, m | 69.7, CH | 3.17, m | 69.7, CH | 3.10, br s | 69.9, CH | 3.12, br s | 69.6, CH |
| 5′′′ | 3.37, m | 77.1, CH | 3.47, m | 77.2, CH | 3.10, br s | 77.5, CH | 3.36, m | 77.0, CH |
| 6′′′ | 3.71, d (9.6); 3.45, m | 61.7, CH2 | 3.73, m; 3.47, d (7.6) | 60.7, CH2 | 3.57, m; 3.35, br s | 60.8, CH2 | 3.67, m; 3.43, m | 60.6, CH2 |
| OCH3 | 3.76, s | 58.1, CH3 | 3.79, s | 59.7, CH3 | 3.85, s | 55.4, CH3 | ||
ε) 265 (3.74) nm; IR (KBr) νmax 3411, 2925, 1681, 1624, 1531, 1141 cm−1; HRESIMS m/z 513.1760 [M + H]+ (calcd for C27H29O10, 513.1755). 1H NMR (400 MHz, DMSO-d6) and 13C NMR (100 MHz, DMSO-d6), see Table 1.ε) 269 (3.27) nm; IR (KBr) νmax 3430, 2926, 1635, 1448, 1074 cm−1; HRESIMS m/z 585.2337 [M + H]+ (calcd for C31H37O11, 585.2330). 1H NMR (400 MHz, DMSO-d6) and 13C NMR (100 MHz, DMSO-d6), see Table 3.
| No. | 8a | 16a | 16b | 16c | ||||
|---|---|---|---|---|---|---|---|---|
| δH (J in Hz) | δC, type | δH (J in Hz) | δC, type | δH (J in Hz) | δC, type | δH (J in Hz) | δC, type | |
| 2 | 8.23, s | 156.2, CH | 4.10, br d (10.0); 3.92, t (10.0) | 69.1, CH2 | 4.11, br d (10.4); 3.88, t (10.4) | 69.4, CH2 | 4.23, br d (10.4); 3.73, t (10.4) | 69.6, CH2 |
| 3 | 121.2, C | 3.32, br s | 30.5, CH | 3.35, br s | 30.4, CH | 3.63, br s | 29.7, CH | |
| 4 | 181.4, C | 2.79, dd (15.6, 4.4); 2.63, m | 26.2, CH2 | 2.80, m; 2.67, m | 26.1, CH2 | 2.60, m | 27.4, CH2 | |
| 5 | 157.9, C | 156.8, C | 156.5, C | 156.8, C | ||||
| 6 | 112.6, C | 113.1, C | 115.4, C | 112.9, C | ||||
| 7 | 160.6, C | 154.4, C | 154.9, C | 154.9, C | ||||
| 8 | 6.79, s | 93.2, CH | 6.10, s | 98.7, CH | 6.39, s | 99.3, CH | 6.09, s | 98.7, CH |
| 9 | 155.5, C | 152.5, C | 152.9, C | 152.8, C | ||||
| 10 | 106.0, C | 107.0, C | 109.8, C | 107.0, C | ||||
| 1′ | 109.4, C | 124.2, C | 119.5, C | 121.4, C | ||||
| 2′ | 154.0, C | 154.9, C | 154.5, C | 154.3, C | ||||
| 3′ | 115.3, C | 118.7, C | 116.0, C | 126.5, C | ||||
| 4′ | 156.4, C | 152.9, C | 153.1, C | 153.0, C | ||||
| 5′ | 6.38, d (8.4) | 106.6, CH | 6.62, d (8.4) | 106.7, CH | 6.34, d (8.4) | 107.2, CH | 6.62, d (8.4) | 111.7, CH |
| 6′ | 6.75, d (8.4) | 128.7, CH | 6.87, d (8.4) | 123.4, CH | 6.74, d (8.4) | 124.0, CH | 6.87, d (8.4) | 124.6, CH |
| 1′′ | 3.46, m; 3.24 m | 21.3, CH2 | 3.14, m | 22.3, CH2 | 3.18, d (6.4) | 22.4, CH2 | 3.15, m | 22.2, CH2 |
| 2′′ | 5.20, m | 122.1, CH | 5.13, t (6.8) | 122.9, CH | 5.15, m | 123.6, CH | 5.13, t (7.2) | 124.0, CH |
| 3′′ | 130.8, C | 129.2, C | 129.3, C | 129.1, C | ||||
| 4′′ | 1.61, s | 25.6, CH3 | 1.61, s | 25.5, CH3 | 1.61, s | 25.5, CH3 | 1.62, s | 25.4, CH3 |
| 5′′ | 1.71, s | 18.7, CH3 | 1.69, s | 17.7, CH3 | 1.71, s | 17.7, CH3 | 1.69, s | 17.6, CH3 |
| 1′′′ | 3.24, m | 22.4, CH2 | 3.52, m; 3.30, br s | 22.6, CH2 | 3.27, d (6.4) | 22.5, CH2 | 3.52, m; 3.30, br s | 23.0, CH2 |
| 2′′′ | 5.20, m | 123.5, CH | 5.18, t (6.8) | 124.2, CH | 5.19, m | 124.2, CH | 5.18, t (7.2) | 124.3, CH |
| 3′′′ | 129.5, C | 129.7, C | 129.6, C | 129.5, C | ||||
| 4′′′ | 1.61, s | 25.6, CH3 | 1.61, s | 25.6, CH3 | 1.61, s | 25.5, CH3 | 1.62, s | 25.5, CH3 |
| 5′′′ | 1.74, s | 17.3, CH3 | 1.73, s | 17.9, CH3 | 1.71, s | 17.8, CH3 | 1.71, s | 18.0, CH3 |
| 1′′′′ | 5.03, d (6.4) | 100.4, CH | 4.71, d (7.6) | 101.3, CH | 4.71, d (6.8) | 101.3, CH | 4.46, d (7.6) | 104.7, CH |
| 2′′′′ | 3.26, m | 73.4, CH | 3.24, m | 73.5, CH | 3.17, br s | 73.4, CH | 3.22, m | 73.8, CH |
| 3′′′′ | 3.26, m | 76.7, CH | 3.24, m | 77.0, CH | 3.17, br s | 76.9, CH | 3.13, m | 76.3, CH |
| 4′′′′ | 3.17, m | 69.7, CH | 3.14, m | 69.8, CH | 3.14 m | 69.8, CH | 2.99, m | 70.1, CH |
| 5′′′′ | 3.46, m | 77.3, CH | 3.32, br s | 77.0, CH | 3.26, m | 77.1, CH | 2.97, m | 77.4, CH |
| 6′′′′ | 3.72, m; 3.44, m | 60.7, CH2 | 3.69, br d (11.2); 3.46, m | 60.0, CH2 | 3.70, d (10.0); 3.43, m | 60.1, CH2 | 3.72, m; 3.35, br s | 61.4, CH2 |
| OCH3 | 3.60, s | 60.8, CH3 | 3.63, s | 60.8, CH3 | 3.57, s | 59.9, CH3 | ||
ε) 261 (3.32) nm; IR (KBr) νmax 3452, 2925, 1656, 1479, 1190, 1052, 835 cm−1; HRESIMS m/z 515.1562 [M + H]+ (calcd for C26H27O11, 515.1548). 1H NMR (400 MHz, DMSO-d6) and 13C NMR (100 MHz, DMSO-d6), see Table 1.ε) 257 (3.17) nm; IR (KBr) νmax 3412, 2919, 1655, 1443, 1207, 1074 cm−1; HRESIMS m/z 537.1369 [M + Na]+ (calcd for C26H26O11Na, 537.1367); 1H NMR (600 MHz, DMSO-d6) and 13C NMR (150 MHz, DMSO-d6), see Table 1.ε) 281 (3.95) nm; IR (KBr) νmax 3411, 2928, 1617, 1077 cm−1; HRESIMS m/z 587.2865 [M + H]+ (calcd for C32H43O10, 587.2851). 1H NMR (400 MHz, DMSO-d6) and 13C NMR (100 MHz, DMSO-d6), see Table 3.ε) 281 (3.95) nm; IR (KBr) νmax 3411, 2921, 1613, 1075, 1039 cm−1; HRESIMS m/z 587.2865 [M + H]+ (calcd for C32H43O10, 587.2851). 1H NMR (400 MHz, DMSO-d6) and 13C NMR (100 MHz, DMSO-d6), see Table 3.ε) 281 (3.95) nm; IR (KBr) νmax 3429, 2923, 1616, 1075, 1028 cm−1; HRESIMS m/z 587.2863 [M + H]+ (calcd for C32H43O10, 587.2851). 1H NMR (400 MHz, DMSO-d6) and 13C NMR (100 MHz, DMSO-d6), see Table 3.ε) 273 (3.24) nm; IR (KBr) νmax 3431, 2923, 1606, 1465, 1075 cm−1; HRESIMS m/z 531.1863 [M + H]+ (calcd for C27H31O11, 531.1863). 1H NMR (400 MHz, DMSO-d6) and 13C NMR (100 MHz, DMSO-d6), see Table 2.ε) 270 (3.76) nm; IR (KBr) νmax 3432, 2922, 2853, 1653, 1609, 1571, 1066, 1027 cm−1; HRESIMS m/z 549.1967 [M + H]+ (calcd for C27H33O12, 549.1966). 1H NMR (400 MHz, DMSO-d6) and 13C NMR (100 MHz, DMSO-d6), see Table 2.ε) 271 (3.05) nm; IR (KBr) νmax 3432, 2922, 1613, 1254, 1072 cm−1; HRESIMS m/z 487.1972 [M + H]+ (calcd for C26H31O9, 487.1963). 1H NMR (400 MHz, DMSO-d6) and 13C NMR (100 MHz, DMSO-d6), see Table 2.Hydrolysis and IC-PAD analysis of 1a, 16a and 22a
To identify the sugar residues, compounds 1a (1.84 mg), 16a (0.84 mg) and 22a (1.10 mg) were hydrolyzed and then analyzed by IC-PAD, as we had previously reported.21Crude enzyme catalysis of 1
Mucor hiemalis was grown in 250 mL Erlenmeyer flasks at room temperature for three days to reach the exponential phase. The mycelia were collected and ground in a mortar with liquid nitrogen. An aliquot of 1 g of the powder was suspended in 1 mL of 50 mM Tris–HCl buffer (pH 8.0) and 100 μL protease inhibitor Cocktail Set IV (Beijing Hanlinyuanzhi Biological Technoloty Co., Ltd., Beijing, China) with agitation at 4 °C. The mixture was centrifuged at 9000 rpm for 30 min at 4 °C. The sediment was treated with 1 mL of Tris–HCl buffer (pH 8.0) containing 500 mM UDPG, 5 mM MgCl2, 20% glycerol, and 100 mM glycycoumarin (1) as the substrate for 24 h at 30 °C. The reaction mixture was extracted with EtOAc and then injected for HPLC analysis.Water solubility test of 1 and 1a
The water solubility test was conducted by the saturation shake-flask method.22 Briefly, the pure solid sample of 1 and 1a was added into 10 mL of neutral aqueous solution in a conical flask, respectively. The solution containing an excess of 1 and 1a was stirred at room temperature for more than 6 hours. After more than 18 hours of sedimentation, the saturated solution were filtered and measured by HPLC.Cytotoxic activity and Nrf2 activation assay
The cytotoxic and Nrf2 activation activities were evaluated as we had previously reported.6,23Evaluation of intestinal absorption of 1 and 1a using Caco-2 cell model
We used the intestinal Caco-2 cell monolayer model to evaluate the intestinal absorption properties of 1 and 1a, as we have recently reported.24Results and discussion
Glucosylation of glycycoumarin by filamentous fungi
Using glycycoumarin (1) as the substrate, we screened 26 strains of filamentous fungi for their capabilities to catalyze the glucosylation reaction. These strains were derived from Absidia, Alternaria, Aspergillus, Botrytis, Crebrothecium, Cunninghamella, Doratomyces, Fusarium, Gibberella, Mucor, Penicillium, Phoma, and Rhizopus species (Table S5†). Fourteen strains could convert 1 into a more polar product 1a ([M − H]− m/z 529) after three days of co-incubation, according to liquid chromatography coupled with mass spectrometry (LC-MS) analysis. In tandem mass spectrometry, the [M − H]− ion could lose 162 Da to yield m/z 367, suggesting it should be an O-glucoside of 1 (Fig. 1). Among these strains, Mucor hiemalis CGMCC 3.14114 was the most potent one, with a conversion rate of 100%. A time-course study indicated that 1 could be almost exhaustively metabolized into 1a within 12 h (Fig. S1†). | ||
| Fig. 1 Glucosylation of glycycoumarin (1) by Mucor hiemalis. (A) The biocatalytic reaction. (B) HPLC chromatograms of 1 and its biotransformed product 1a. (C) (−)-ESI-MS and MS/MS spectra for 1a. | ||
A scaled-up biotransformation of 1 in M. hiemalis for three days was conducted, and 1a was purified by column chromatography and semi-preparative liquid chromatography. The HRESIMS spectrum established the molecular formula of 1a as C27H30O11 ([M + H]+ m/z 531.1856, calcd for C27H31O11, 531.1861). The 1H and 13C NMR spectra showed additional resonances corresponding to a glucosyl residue (δH 4.98, d, J = 7.8 Hz; δC 100.7, 77.2, 76.8, 73.4, 69.8 and 60.8). The δH 4.98 and δC 100.7 signals could be assigned to the anomeric H-1′′′ and C-1′′′, respectively, according to the HSQC spectrum. H-1′′′ showed a long-range correlation with δC 158.3 (C-7), indicating the glucosyl residue was linked to 7-OH (instead of 2′- or 4′-OH) of 1. In accordance, the resonance for C-7 shifted upfield by 1.8 ppm, whereas C-6 and C-10 shifted downfield by 1.7 and 1.8 ppm, respectively, when compared to 1 (Table 1). The coupling constant of H-1′′′ (7.8 Hz) suggested β-configuration of the glycosidic bond. To further confirm the glucosyl residue, 1a was subjected to acid hydrolysis, and then analyzed by ion chromatography coupled with pulsed amperometric detection (IC-PAD).21 The hydrolysis product showed a peak at the same retention time as D-glucose (Fig. S2†). On the basis of the above deductions, 1a was identified as glycycoumarin 7-O-β-D-glucoside, which is a new compound.
Substrate promiscuity of glucosylation catalyzed by Mucor hiemalis
The above results indicated that M. hiemalis could efficiently and selectively catalyze the glucosylation of glycycoumarin (1) at 7-OH, which is neighboring to the isoprenyl group. Unexpectedly, this reaction did not take place at 2′-OH or 4′-OH, which had relatively low steric hindrance. Thus, the isoprenyl group may be critical for this reaction. In order to explore the catalytic promiscuity, we tested the biotransformation of 28 prenylated phenolic compounds by M. hiemalis (Fig. 2). These compounds contain different basic skeletons, including 3-aryl coumarin (1–3), pterocarpan (4), isoflavone (5–14), isoflavanone (15), isoflavan (16, 17), flavonol (18–22), coumarone (23), flavanone (24, 25), chalcone (26, 27), and others (28). The biotransformed products were analyzed by LC-UV/MS (Fig. S3–S30†). Interestingly, all the 28 compounds could be glycosylated, and the conversion rates were above 90% (except for 3 and 22). Hydroxylation (+16 Da) and sulfation (+80 Da) were observed as side reactions for a few compounds (3, 4, 18, 21, 27), though the conversion rates were very low (Table S6†).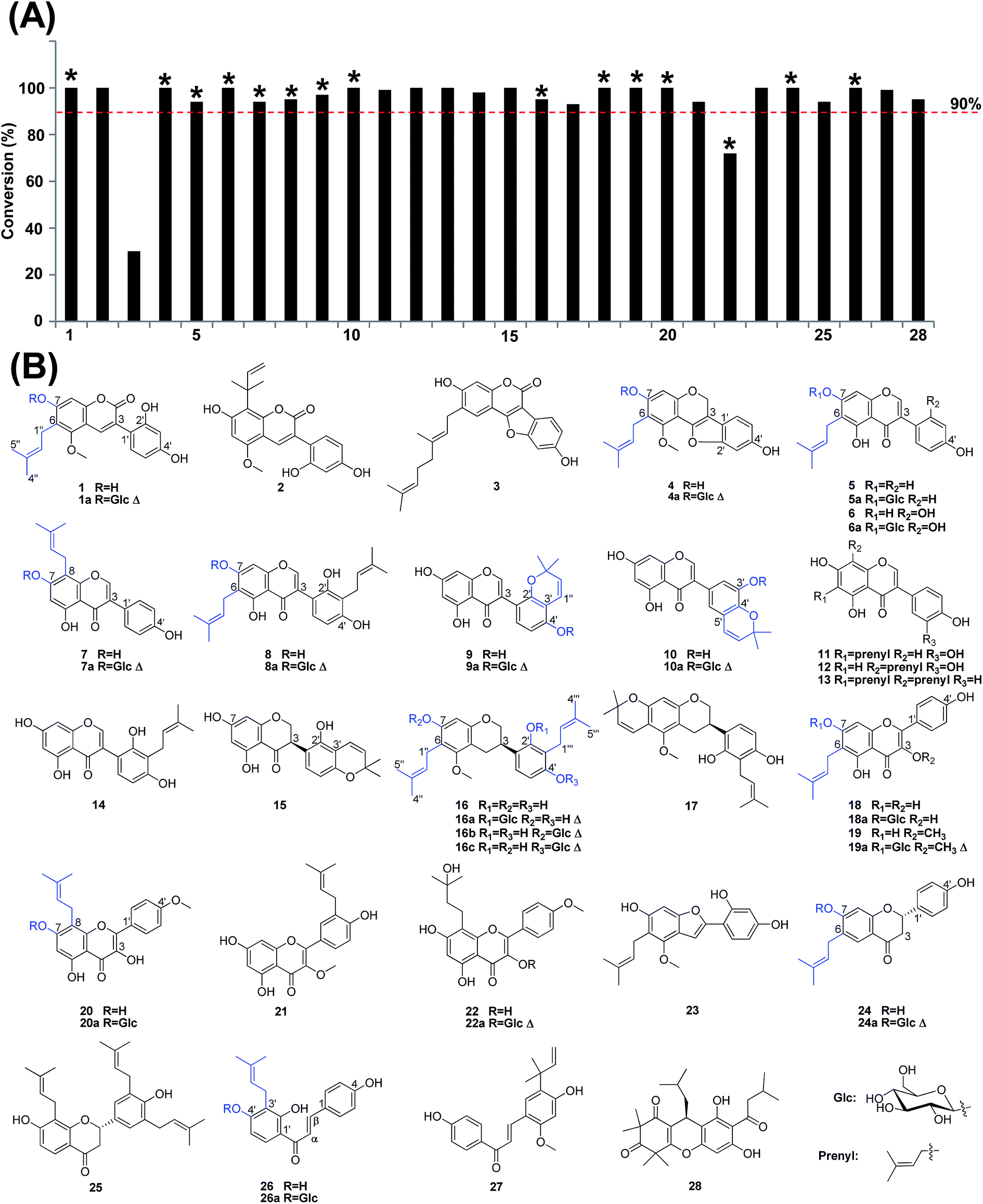 | ||
| Fig. 2 Exploring the catalytic promiscuity of Mucor hiemalis. (A) Total conversion rates (%) for substrates 1–28. (B) Structures of the substrates and their glycosylated products. The asterisk (*) indicates that the products were prepared by scaled-up biotransformation and identified by MS and NMR spectroscopy; the triangle (Δ) indicates new compounds. Compounds identification: 1, glycycoumarin; 2, licoarylcoumarin; 3, puerarol; 4, glyurallin A; 5, wighteone; 6, luteone; 7, lupiwighteone; 8, angustone A; 9, allolicoisoflavone B; 10, semilicoisoflavone B; 11, 6-C-prenylorohol; 12, gancaonin L; 13, 6,8-diprenylgenistein; 14, licoisoflavone A; 15, licoisoflavanone; 16, licoricidin; 17, cyclized licoricidin; 18, licoflavonol; 19, topazolin; 20, icaritin; 21, 5,7,4′-trihydroxy-3′-(3-methylbut-2-enyl)-3-methoxy flavone; 22, wushanicaritin; 23, licocoumarone; 24, bavachin; 25, sophoranone; 26, corylifolinin; 27, licochalcone A; 28, rhodomyrtone. | ||
Because all the above substrates contain more than one hydroxyl group, it is essential to determine the glycosylation position. Thus, we conducted scaled-up biotransformation of 15 substrates, and obtained 17 mono-O-glucosylated products. Twelve of them are new compounds, and their structures were fully identified by HRESIMS and NMR spectroscopic analysis. The glucosyl residue exhibited characteristic NMR resonances (the anomeric proton signal at δH 4.46–5.50; the anomeric carbon signal at δC 100.1–104.7; one CH2 signal at δC 60.0–61.7; and four CH signals at δC 69.6–77.5). The coupling constants of the anomeric protons (J = 6.8–8.4 Hz) suggested β-glycosidic linkage for all the products. The glucosyl residue for 16a and 22a was further confirmed by IC-PAD analysis after acid hydrolysis (Fig. S38†). The linkage site for glucosyl residue was determined by HBMC correlations. 13C and 1H NMR spectral data for the new compounds are given in Tables 1–3.
Here we describe structural identification of 16a–16c as typical examples. The scaled-up biotransformation of 16 by M. hiemalis yielded three glycosylated products, namely 16a–16c. They had the same molecular formula C32H42O10, according to their HRESIMS spectra. In the MS/MS spectra, all the [M − H]− ions (m/z 585) could lose 162 Da to yield m/z 423 (Fig. S18†), suggesting they were mono-O-glucosides. In the HMBC spectra, the anomeric proton H-1′′′ showed a long-range correlation with δC 154.9 (C-2′, 16a), δC 154.9 (C-7, 16b), and δC 153.0 (C-4′, 16c), respectively, indicating the glucosyl residue was linked to 2′-OH of 16a, 7-OH of 16b, and 4′-OH of 16c. In accordance, the ortho-carbons resonated at remarkably low fields when compared to 16. For instance, C-1′ (δC 124.2) and C-3′ (δC 118.7) of 16a shifted down-field by 4.5 and 2.8 ppm, respectively (Table S4†). Based on the above deductions, the structures for 16a–16c were identified as licoricidin 2′-O-β-D-glucoside, licoricidin 7-O-β-D-glucoside, and licoricidin 4′-O-β-D-glucoside, respectively. All of them are new compounds.
Through the above structural elucidations, we found that all the prenylated phenolic compounds could be efficiently glucosylated at the isoprenyl-neighboring hydroxyl group (Fig. 2). This rule applied when the isoprenyl group was located at C-6 (1, 4–6, 18, 19, 24) or C-8 (7, 20, 26) (please be noted that carbon numbering for chalcones is different from the other subclasses of flavonoids). For both groups of compounds (C-6 or C-8), 7-O-glucoside was the only or predominant product, though other free hydroxyl groups are also present in the substrate structures. It was important to note that compounds 5, 6, 18 and 19 contained two isoprenyl-neighboring hydroxyl groups (5-OH and 7-OH) in their structures, and all of them were only glucosylated at 7-OH. This was probably due to the intramolecular hydrogen bonding between 5-OH and the carbonyl group at C-4. Compound 16 contains two isoprenyl groups at C-6 and C-3′, and yielded three glucosides at 7-OH, 2′-OH, and 4′-OH. Compound 8 also contains two isoprenyl groups, and yielded 7-O-glucoside as the predominant product. The three minor products could only be observed by LC-MS analysis (Fig. S10†). The above evidences indicated that glucosylation occurred more readily at 7-OH than at 2′- or 4′-OH. Aside from the normal isoprenyl (γ,γ-dimethylallyl) chain, a cyclized isoprenyl group (such as a 2,2-dimethylbenzopyran ring) could also facilitate the glucosylation reaction, as exemplified by allolicoisoflavone B (9) and semilicoisoflavone (10). Licoarylcoumarin (2), licochalcone A (27) and rhodomyrtone (28) contain varied isoprenyl groups (α,α-dimethylallyl- or 3-methyl-1-oxo-butyl-). All of them could be efficiently glucosylated with conversion rates of above 95%. The two isoprenyl chains of 6,8-diprenylgenistein (13) did not hinder the glucosylation reaction. In fact, it was completely metabolized into one single product, which was expected to be the 7-O-glucoside. Puerarol (3) contains a C10 monoterpene group (two isoprenyl units). It could also be metabolized into O-glucosides, though the conversion rate decreased to 30%. Compound 22 contains a hydroxylated isoprenyl group (3-hydroxy-3-methyl-butyl-). Although it could also be efficiently glycosylated, the product was identified as 3-O-glucoside. This special isoprenyl group may suppress 7-O-glucosylation.
We further tested seven other phenolic compounds with no isoprenyl groups (29–35). Only the two flavonols, kaempferol (29) and quercetin (30), could be glycosylated (Table S6, Fig. S31–S37†). Their products were obtained by scaled-up biotransformation. Interestingly, both products (29a and 30a) were 3-O-glucosides. This result indicated that M. hiemalis could catalyze the glucosylation of 3-OH of flavonols, when no isoprenyl group was present. In a previous study, M. hiemalis had also been reported to catalyze the glucosylation of normal phenolic compounds.25
Although prenylated phenolics are common in natural products, only a few of them are present as glycosides.26 Glycyrrhiza uralensis contains tens of prenylated flavonoids and flavonoid glycosides. However, no compounds reported from this plant, thus far, contain both an isoprenyl group and a sugar residue. The present work provides an efficient approach to prepare the glycosides of prenylated phenolic compounds, and the reaction took place specifically at the isoprenyl-neighboring hydroxyl group. Our further experiments demonstrated the glucosylation of 1 could also be catalyzed by extracted crude enzymes from M. hiemalis in Tris–HCl buffer, when uridine diphosphate glucose (UDP-G) was added. After optimization of incubating time, donor concentration, pH value, incubating temperature, activating factor, protease inhibitor, and incubating solvents, the conversion rate could be as high as 81.5% (Fig. S39†). A number of glycosyl transferases have been cloned from plants and microorganism, so far.27 However, few reports are available on glycosyl transferases from filamentous fungi. Molecular cloning of this type of enzymes from M. hiemalis is still under way in our laboratory.
Effects of glycosylation on solubility, intestinal absorption, and bioactivities
We tested the solubility of 1 and 1a in water by the saturation shake-flask method.22 Compound 1 could hardly be dissolved in water, while 1a had a solubility of 130 mg L−1 in water at room temperature (Table S7†). This result indicated that glycosylation could significantly enhance water solubility of prenylated phenolic compounds. We also investigated the permeability of 1 and 1a through the intestinal Caco-2 cell monolayer, which is popularly used as an in vitro model to mimic drug intestinal absorption in human. The results revealed that 1a could pass the monolayer remarkably more easily than 1 (PAB value, 3.65 × 10−5 cm s−1 for 1a vs. 1.34 × 10−7 cm s−1 for 1), indicating that glycosylation could significantly enhance intestinal absorption (Fig. 3).28 We had reported that flavonoid glycosides tended to be metabolized into their aglycones after oral administration.29,30 Based on the above results, the glucosyl residue may be able to function as a carrier to introduce insoluble and poorly bioavailable compounds into circulation.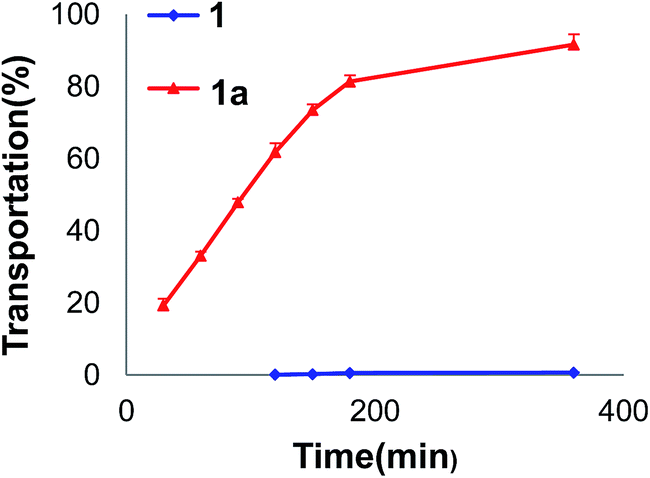 | ||
| Fig. 3 The time-transport rate curves of 1 and 1a from the apical (AP) to basolateral (BL) side in the Caco-2 monolayer cell model. | ||
A lot of prenylated phenolic compounds show significant cytotoxic activities.1 Thus, we tested the cytotoxic activities of several substrates (5, 6, 8–10, 16, 18, 19, 24, 26), together with their glycosides, against HepG2 human hepatocellular carcinoma cell line at 10 μM using the MTS assay. For majority of the tested compounds, the cytotoxic activities decreased remarkably after being glycosylated (Fig. S40†). Another test indicated the glycosides exhibited comparable Nrf2 activation activities to the substrates (Fig. S41†).23 More bioactivity tests, including the hepatoprotective activities of 1a (so as to compare its activities with glycycoumarin), will be conducted in the near future.7
Conclusions
In conclusion, this work reports a highly efficient and regio-specific biocatalytic method to prepare the O-glucosides of prenylated phenolic compounds, which are rare in natural products. Mucor hiemalis could selectively catalyze glucosylation of the isoprenyl-neighboring hydroxyl group, rather than other hydroxyls with low steric hindrance. Scaled-up biotransformation of 15 compounds yielded 17 products, and 12 of them are new compounds. Glycosylation could significantly enhance water solubility and intestinal absorption of these lipophilic compounds, and thus increase their druggability. The glycosylation reaction is fast, efficient, and environment-friendly. Molecular cloning of the glycosyl transferase catalyzing this reaction is ongoing in our laboratory.Acknowledgements
This work was supported by National Natural Science Foundation of China (No. 81173644, No. 81222054).References
- M. Marín and S. Máňez, Curr. Med. Chem., 2013, 20, 272–279 CrossRef.
- B. Botta, A. Vitali, P. Menendez, D. Misiti and G. D. Monache, Curr. Med. Chem., 2005, 12, 713–739 CrossRef CAS.
- D. Barron and R. K. Ibrahim, Phytochemistry, 1996, 43, 921–982 CrossRef CAS.
- Q. Wang, W. J. Miao, C. Xiang, D. A. Guo and M. Ye, Chin. Tradit. Herb. Drugs, 2012, 43, 1886–1890 CAS.
- Q. Wang, W. J. Miao, C. Xiang, D. A. Guo and M. Ye, Chin. Tradit. Herb. Drugs, 2014, 45, 31–36 CAS.
- (a) S. Ji, X. Qiao, Z. W. Li, Y. R. Wang, S. W. Yu, W. F. Liang, X. H. Lin and M. Ye, RSC Adv., 2015, 5, 45258–45265 RSC; (b) S. Ji, Z. W. Li, W. Song, Y. R. Wang, W. F. Liang, K. Li, S. N. Tang, Q. Wang, X. Qiao, D. M. Zhou, S. W. Yu and M. Ye, J. Nat. Prod., 2016 DOI:10.1021/acs.jnatprod.5b00877.
- X. H. Song, S. T. Yin, Y. Z. Huo, M. Liang, L. H. Fan, M. Ye and H. B. Hu, Free Radical Biol. Med., 2015, 89, 135–146 CrossRef CAS PubMed.
- M. Adianti, C. Aoki, M. Komoto, L. Deng, I. Shoji, T. S. Wahyuni, M. I. Lusida, Soetjipto, H. Fuchino, N. Kawahara and H. Hotta, Microbiol. Immunol., 2014, 58, 180–187 CrossRef CAS PubMed.
- Eerdunbayaer, M. A. A. Orabi, H. Aoyama, T. Kuroda and T. Hatano, Molecules, 2014, 19, 3883–3897 CrossRef CAS PubMed.
- G. J. Williams, C. S. Zhang and J. S. Thorson, Nat. Chem. Biol., 2007, 3, 657–662 CrossRef CAS PubMed.
- B. Yu, J. S. Sun and X. Y. Yang, Acc. Chem. Res., 2012, 45, 1227–1236 CrossRef CAS PubMed.
- Y. Yang, X. H. Zhang and B. Yu, Nat. Prod. Rep., 2015, 32, 1331–1355 RSC.
- J. B. Xiao, T. S. Muzashvili and M. I. Georgiev, Biotechnol. Adv., 2014, 32, 1145–1156 CrossRef CAS PubMed.
- L. Pistelli, A. Bertoli, I. Giachi and A. Manunta, J. Nat. Prod., 1998, 61, 1404–1406 CrossRef CAS PubMed.
- Y. Shibuya, S. Tahara, Y. Kimura and J. Z. Mizutani, Naturforscher, 1991, 46, 513–518 CAS.
- V. Reutrakul, N. Ningnuek, M. Pohmakotr, C. Yoosook, C. Napaswad, J. Kasisit, T. Santisuk and P. Tuchinda, Planta Med., 2007, 73, 683–688 CrossRef CAS PubMed.
- G. Wang, C. Y. Wang, B. J. Zhang, Y. Tian, S. G. Tan, T. X. Liu, D. Sa, J. Hou, J. Y. Peng, J. H. Yao and X. C. Ma, J. Liq. Chromatogr. Relat. Technol., 2013, 36, 1163–1176 CAS.
- G. Cioffi, L. M. Escobar, A. Braca and N. D. Tommasi, J. Nat. Prod., 2003, 66, 1061–1064 CrossRef CAS PubMed.
- Y. Wei, Q. Q. Xie, D. Fisher and I. A. Sutherland, J. Chromatogr. A, 2011, 1218, 6206–6211 CrossRef CAS PubMed.
- G. Ye, C. Li, C. G. Huang, Z. X. Li, X. L. Wang and Y. Z. Chen, China J. Chin. Mater. Med., 2005, 30, 1340–1342 Search PubMed.
- W. Song, L. L. Si, S. Ji, H. Wang, X. M. Fang, L. Y. Yu, R. Y. Li, L. N. Liang, D. M. Zhou and M. Ye, J. Nat. Prod., 2014, 77, 1632–1643 CrossRef CAS PubMed.
- B. Edit, E. A. C. John and T. Krisztina, J. Pharm. Biomed. Anal., 2008, 46, 335–341 CrossRef PubMed.
- (a) W. F. Liang, Z. W. Li, S. Ji, Q. Wang, X. Qiao, D. A. Guo and M. Ye, RSC Adv., 2015, 5, 63753–63756 RSC; (b) X. H. Lin, M. N. Cao, W. N. He, S. W. Yu, D. A. Guo and M. Ye, Phytochemistry, 2014, 105, 129–134 CrossRef CAS PubMed.
- Q. Wang, X. Qiao, Y. Qian, Z. W. Li, Y. M. Tzeng, D. M. Zhou, D. A. Guo and M. Ye, Nat. Prod. Bioprospect., 2015, 5, 237–246 CrossRef CAS PubMed.
- C. Moussa, P. Houziaux, B. Danree and R. Azerad, Drug Metab. Dispos., 1997, 25, 301–310 CAS.
- (a) Y. Koyama, M. Yamamoto, K. Matsunami, H. Otsuka, T. Shinzato and Y. Takeda, J. Nat. Med., 2011, 65, 212–216 CrossRef CAS PubMed; (b) T. Řezanka, J. Jáchymová and V. M. Dembitsky, Phytochemistry, 2003, 62, 607–612 CrossRef; (c) G. Cioffi, L. M. Escobar, A. Braca and D. N. Tommasi, J. Nat. Prod., 2003, 66, 1061–1064 CrossRef CAS PubMed; (d) T. Fukai and T. Nomura, Phytochemistry, 1988, 27, 259–266 CrossRef CAS.
- (a) P. Bojarová, R. R. Rosencrantz, L. Elling and V. Křen, Chem. Soc. Rev., 2013, 42, 4774–4797 RSC; (b) J. B. Xiao, T. S. Muzashvili and M. I. Georgiev, Biotechnol. Adv., 2014, 32, 1145–1156 CrossRef CAS PubMed.
- P. Artursson, K. Palm and K. Luthman, Adv. Drug Delivery Rev., 2012, 64, 280–289 CrossRef.
- C. Xiang, X. Qiao, Q. Wang, R. Li, W. Miao, D. Guo and M. Ye, Drug Metab. Dispos., 2011, 39, 1597–1608 CrossRef CAS PubMed.
- X. Qiao, M. Ye, C. Xiang, Q. Wang, C. Liu, W. Miao and D. Guo, J. Chromatogr. A, 2012, 1258, 84–93 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: LC-MS analysis of the biotransformed products; cytotoxic and Nrf2 activation activities; spectroscopic data (NMR, MS, UV and IR) for new compounds. See DOI: 10.1039/c6ra00072j |
| ‡ These two authors contributed equally to this paper. |
| This journal is © The Royal Society of Chemistry 2016 |