
PhSeBr mediated hydroxylative oxidative dearomatization of naphthols – an open air facile one-pot synthesis of ketols†
Abstract
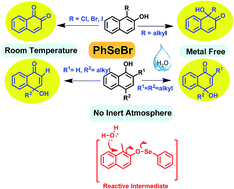
A new methodology for oxidative-dearomatization of planar phenols is described. An economic, viable one-pot metal free protocol for direct conversion of naphthols to α-ketols is reported. Naphthols were found to undergo facile unprecedented oxidative dearomatization with regioselective hydroxylation with phenyl selenyl bromide in open air conditions. Quaternary stereocenters were developed along with formation of sterically demanding α- and γ-ketols with high yields. Functional group tolerance like esters is revealed. A thorough study of the stereoelectronic demands of the unusual oxy-selenium reactive intermediate involved in dearomatization of 1- and 2-naphthols is carried out. 4-Hydroxy cyclohexadieneone and cyclohexadieneone aryl ethers were generated from dialkyl-phenols under similar reaction conditions providing direct evidence of the mechanical postulate. The first instance of the phenoxy–selenium interaction leading to facile dearomatization of arenes is highlighted in this manuscript.
 Please wait while we load your content...
Please wait while we load your content...

