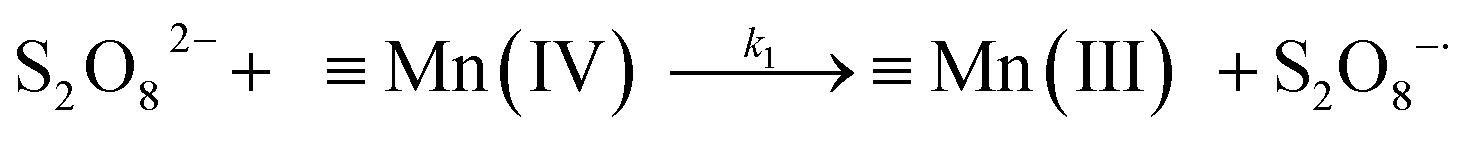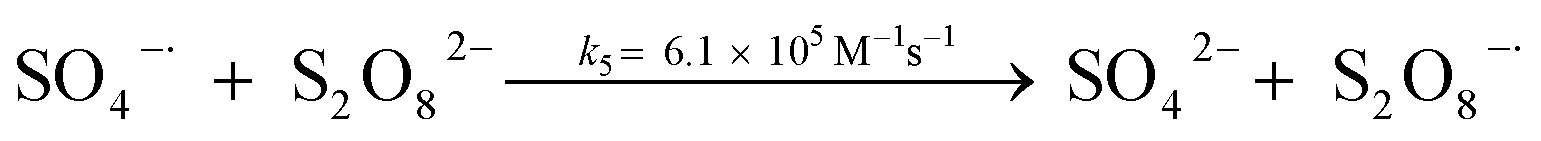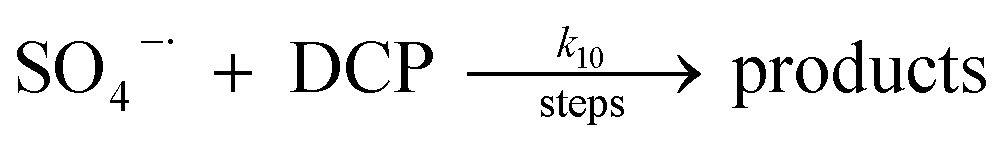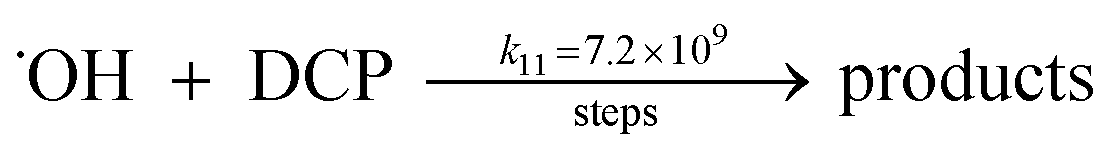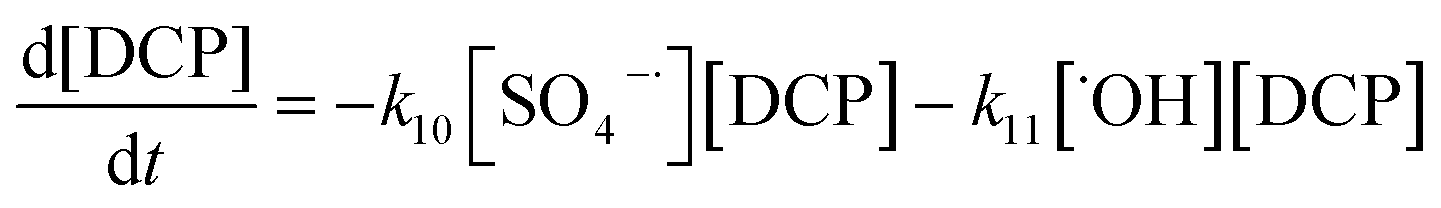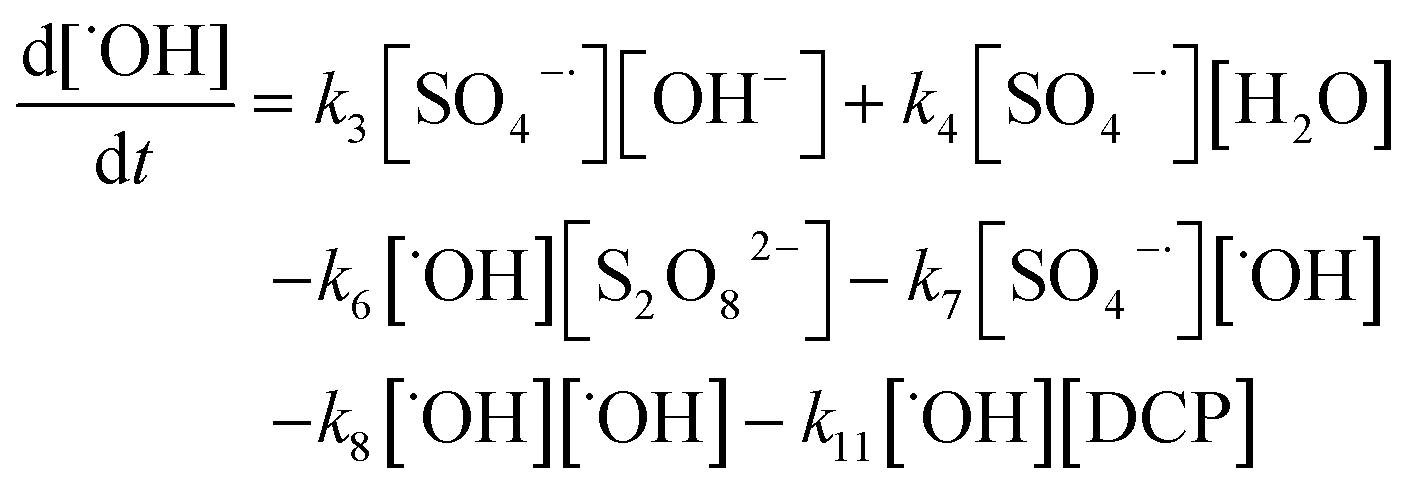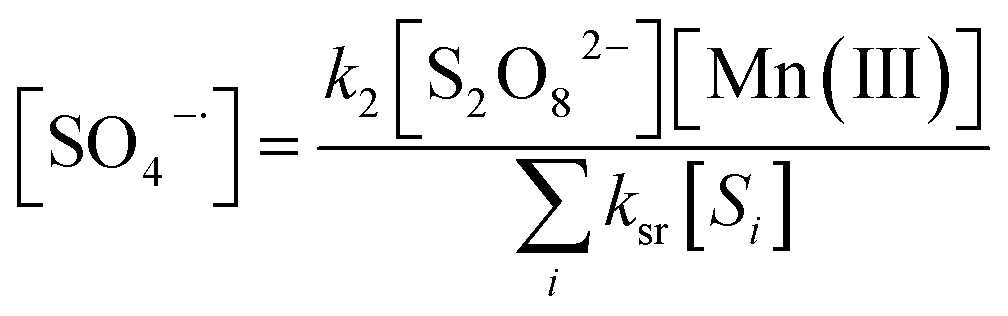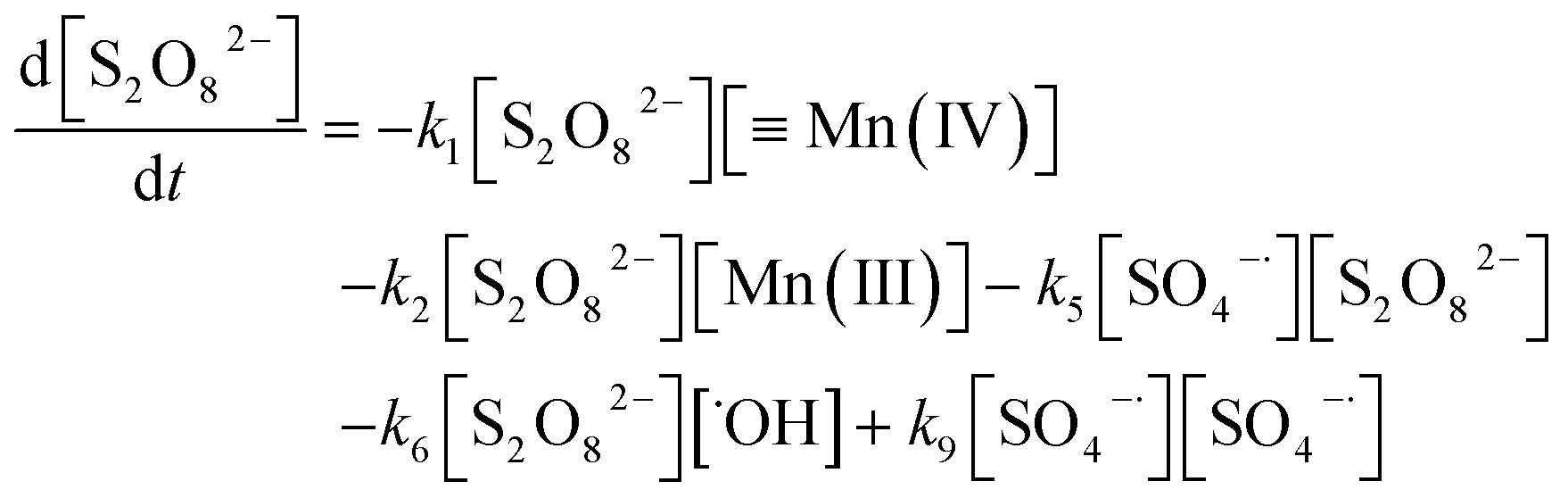DOI:
10.1039/C6RA00008H
(Paper)
RSC Adv., 2016,
6, 35441-35448
Insights into the degradation of 2,4-dichlorophenol in aqueous solution by α-MnO2 nanowire activated persulfate: catalytic performance and kinetic modeling†
Received
1st January 2016
, Accepted 30th March 2016
First published on 31st March 2016
Abstract
In this study, α-MnO2 nanowires were synthesized in a hydrothermal process. These nanowires efficiently activated persulfate (PS) for 2,4-dichlorophenol (DCP) oxidation. The shape and structure of the material were characterized through X-ray diffraction, high-resolution transmission electron microscopy, and energy dispersive X-ray spectroscopy. Quenching tests and electron paramagnetic resonance were conducted, and the results revealed that both ˙OH and SO4˙− were responsible for the degradation of 2,4-DCP in the α-MnO2-activated PS system. A novel kinetic model was established to describe the deterioration of the contaminant under the conditions described in this paper. Several key parameters are investigated in the evaluation process of the kinetic study, including catalyst dosage, PS concentration, pH, and temperature. The maximum removal efficiency of 2,4-DCP was 90.2% at 20.0 mM PS, given 0.2 g L−1 α-MnO2 nanowires, and under a temperature of 30.0 °C. In addition, the α-MnO2 nanowires exhibited relatively stable catalytic activity after five instances of reuse. The aforementioned results indicate that α-MnO2 nanowires are promising catalysts for the activation of PS to degrade organic contaminants in wastewater.
Introduction
Chlorophenols are typically used as preservative agents for dyes, drugs, pesticides, and fungicides and have been listed as priority toxic pollutants by the United States Environmental Protection Agency.1,2 As one of the chlorophenols, 2,4-dichlorophenol (2,4-DCP) in wastewater is a severe global concern because of the persistence, toxicity, and carcinogenicity of this compound.3 Therefore, techniques must be developed for its disposal.
Many efforts have been devoted to developing treatments for 2,4-DCP-contaminated water. Such treatments include physical,4 chemical,3 and biological5,6 methods. However, biodegradation processes usually take relatively long time and the removal efficiency is low, especially at the later stage because of the toxicity of 2,4-DCP to the organisms.7 And physical methods cannot reach the goal to mineralize the 2,4-DCP to small molecular substances.2 Persulfate(PS)-based advanced oxidation processes (AOPs) have been suggested as promising remediation technologies to treat recalcitrant organic contaminants given the high solubility, strong chemical stability, and high redox potential (Eh = 2.01 V) of persulfate (PS).8–10 Considerable effort has been exerted to activate PS with transition metals, heat, alkalinity, and UV;11 among these methods, activation with transition metals (including ions, Co2+, and Ag+) is the most effective. Nevertheless, dissolved metal catalysts are harmful to the environment and are difficult to recover,12,13 thereby limiting the application of this homogeneous system. Under such circumstances, researchers have diverted their attention to the development of heterogeneous catalytic systems that use nanomaterials as an alternative.
MnO2 materials have recently attracted significant interest because the materials have several different phases, including α-, β-, γ-, δ-, ε-, and η- and exhibit various potential as heterogeneous catalysts. Studies have been conducted on the catalytic performance of MnO2 nanomaterials in a Fenton-like reaction to eliminate pollutants from wastewater. Zhang et al. investigated the oxidative removal of methylene blue by using β-MnO2 nanorods in the presence of H2O2.14 Xu et al. evaluated the degradation of steroid estrogens and determined that MnO2 is a promising catalyst to remove estrogens from water.15 Edy et al. synthesized different MnO2 crystallographic phases and tested their capability to facilitate the heterogeneous activation of peroxymonosulfate for phenol degradation.16 However, to the best of our knowledge, no study has been presented regarding the degradation of 2,4-DCP by MnO2 as a catalyst in an advanced oxidation system. Moreover, substantial work must be conducted to identify the reaction kinetics and degradation mechanism of this system given that the implementation of a fundamental kinetic model in a reaction system facilitates the establishment of favourable conditions.
The main objective of this study is to develop a α-MnO2-catalyzed PS AOP to degrade 2,4-DCP in aqueous systems as well as to establish a kinetic model from a mathematical perspective to explain the degradation mechanism. Several key parameters are investigated in the evaluation process of the kinetic study, including catalyst dosage, PS concentration, pH, and temperature. In addition, the dominant radicals that contribute to the process are identified and catalyst reuse examined.
Experimental
Materials
Analytical grade sodium PS (Na2S2O8, 99%), potassium permanganate (KMnO4, 99.5%), acetic acid (CH3COOH, 99.5%), methanol (CH3OH, 99%, MA), tert-butyl alcohol (C4H10O, 98%, TBA), 1,4-benzoquinone (C6H4O2, 98%, BQ), sodium hydroxide (NaOH, 99%), and hydrochloric acid (HCl, 36–38%) were all purchased from Sinopharm Chemical Reagent Co., Ltd. 2,4-DCP (99%) was obtained from Sigma-Aldrich Co., LLC. All the reagents were used without further purification, and solutions were prepared with ultrapure water that was purified by the Milli-Q system to eliminate the influence of water ions on the degradation of 2,4-DCP.
Preparation of the catalyst
The catalyst α-MnO2 was synthesized through a simple modified hydrothermal process.11 In a typical experiment, 0.16 g of KMnO4 was first dispersed in 40 mL of ultrapure water by ultrasonication, and 0.7 mL of acetic acid was then added gradually dropwise. The mixture was transferred to a Teflon-lined autoclave (50 mL) and sealed for heating at 140 °C for 12 h. The obtained products were washed a few times with ultrapure water and ethanol and dried in a vacuum at 60 °C for 8 h. By this method, about 0.05 g α-MnO2 can be collected for one time, then these dry black powders from several preparations were grinded and mixed uniformity for the following experiments.
Experimental procedure
Batch experiments were conducted in 100 mL serum bottles containing 30 mL of the reaction solution. Subsequently, these bottles were immersed in a shaking water bath at 150 rpm. Stock solutions of 2,4-DCP (400 mg L−1) were prepared in ultrapure water prior to each batch experiment. In a typical procedure, a certain amount of catalysts and 2,4-DCP stock solutions were added into the bottles. Then, the reaction was initiated immediately by the addition of PS. At selected time intervals (10, 20, 40, 60, 90, 120, 150, and 180 min), 0.2 mL of the sample was collected from each replicate bottle, and excess methanol was incorporated to quench the reactions of sulfate and hydroxyl radicals.17 Subsequently, the mixed solutions were passed through a 0.22 μm membrane filter for the sampling analysis. The influence of four affecting factors (PS dosage, catalyst dosage, temperature, and pH) on the oxidation of 2,4-DCP was determined. Comparative experiments (without PS or catalysts) were also performed under identical reaction conditions. Each test was conducted in triplicate, and the averaged experimental values were considered. The error bars in the figures represent ± one standard deviation from the mean of the triplicate data.
Analytical methods
The residual 2,4-DCP was quantified through a colorimetric method.18 Aliquots of 600 μL of each sample were mixed with 100 μL of 4-aminoantypirine (20.8 mM), 100 μL of potassium ferricyanide (83.4 mM), and 200 μL of sodium bicarbonate (0.25 M/pH 8.4). After 10 min of coloration, the concentrations of the 2,4-DCP in the reaction mixture at different reaction times were identified by measuring the absorption intensity at λmax = 510 nm.
X-ray diffraction (XRD) patterns were obtained on a Rigaku Smart Lab (3) diffractometer using filtered Cu Kα radiation (λ = 0.1541 nm) with an accelerating voltage of 40 kV and a current of 30 mA. High-resolution transmission electron microscopy (HRTEM) was performed on sample suspensions dried on a Cu coated grid with a Tecnai G2 F20 S-TWIN transmission electron microscope operated at 200 kV.
Electron paramagnetic resonance (EPR) was conducted with 5,5-dimethyl-1-pyrroline N-oxide (DMPO) as a spin-trapping agent to characterize the generation of radicals. Chemical solutions of the DMPO (0.1 M), PS, and catalysts were mixed for 20 seconds and transferred to a 100 μL capillary tube. Then, the capillary tube was inserted into the cavity of the EPR spectrometer (Bruker A200 ESP 300E instrument). The EPR spectrometer was operated under the following conditions: a central field of 323.3 mT, a sweep width of 10 mT, a microwave frequency of 9056 MHz, a microwave power of 0.998 mW, a temperature of 303.0 K, a modulation amplitude of 600, and a sweep time of 60 seconds.
Results and discussion
Characterization of the α-MnO2 catalyst
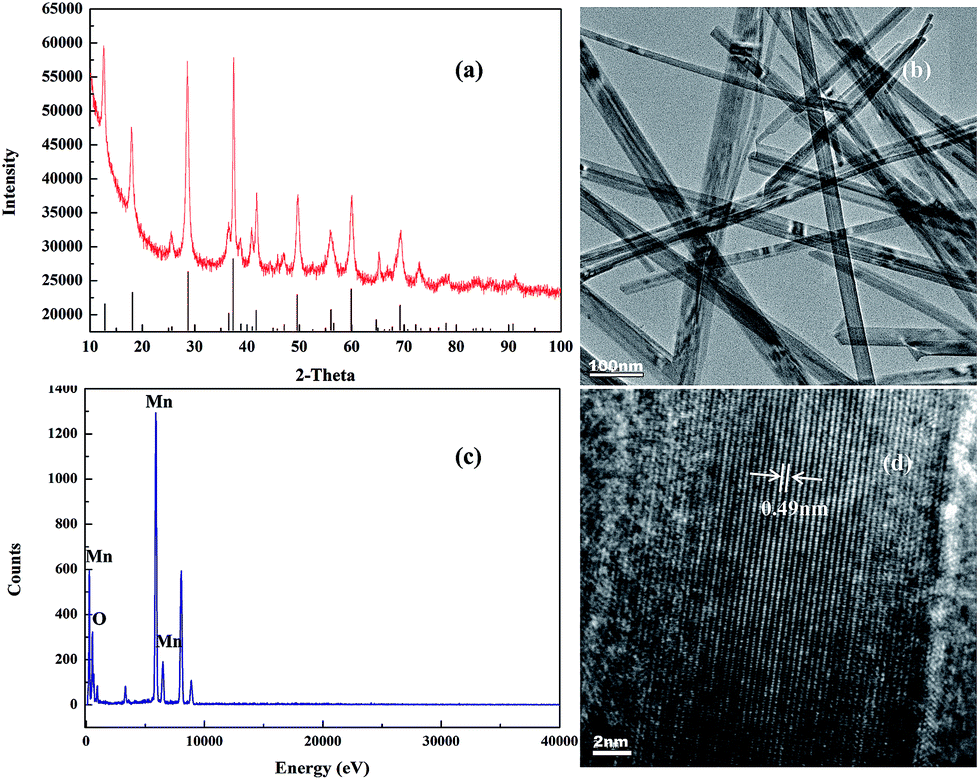
The structures of the synthetic catalyst were examined through X-ray diffraction (XRD). As shown in Fig. 1(a), the sample exhibits crystalline peaks at 2θ = 12.8°, 18.1°, 28.6°, and 37.5° in the angle range of 10–90°. These peaks conform to the characteristic peaks of α-MnO2 as reported in (JPCD 44-0141).19,20 Fig. 1(b) and (d) depict the transmission electron microscopy (TEM) and high-resolution TEM (HRTEM) images of synthetic α-MnO2, which takes the form of well-defined nanowires with a diameter of 10–50 nm. Lattice fringes were clearly visible with a spacing of 0.49 nm, which is consistent with the spacing of α-MnO2 planes.11 Fig. 1(c) is energy dispersive X-ray spectra (EDX), it shows that the catalyst contains Mn and O. The test report shows the atomic percentage of Mn and O in the composites are 30.83% and 60.16%, which is almost equal to the molar ratio of MnO2.
 |
| | Fig. 1 Characterization of the catalyst. (a) X-ray diffraction pattern; (b) transmission electron microscopy (TEM) image; (c) energy dispersive X-ray spectra of the composites; and (d) high-resolution TEM image. | |
Comparison of 2,4-DCP degradation in different systems
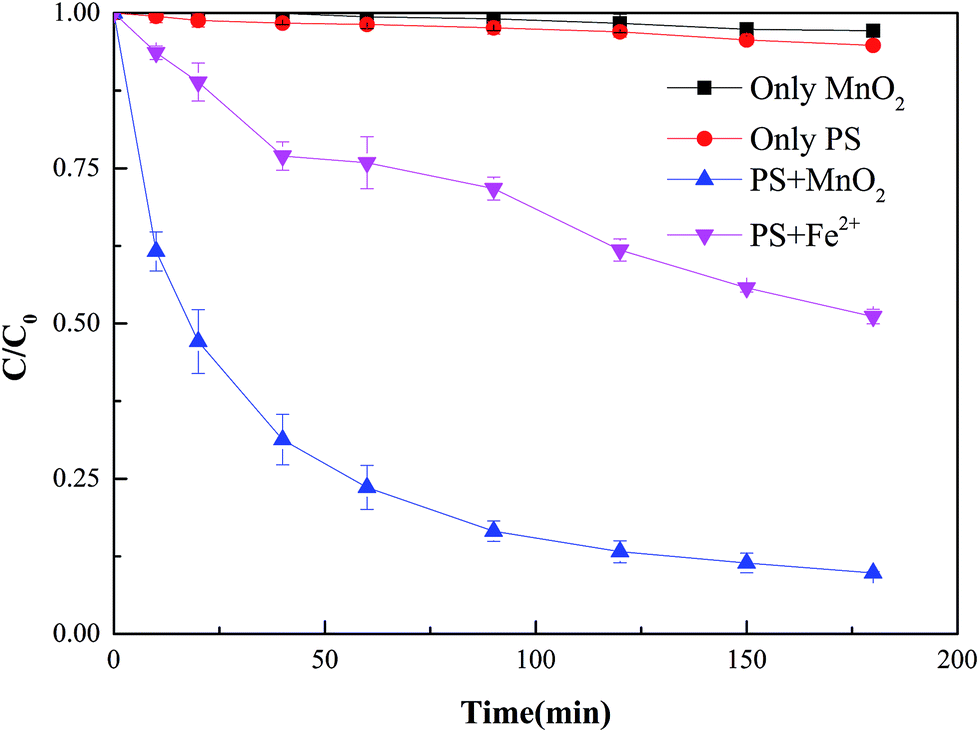
To determine the effect of synthesized α-MnO2 nanowires on PS activation in the degradation process of 2,4-DCP, comparative experiments were performed on PS, α-MnO2 nanowires, PS/α-MnO2 nanowires, and PS/Fe2+ systems. Operations were initiated in the same reactor and controlled under the same conditions. The variation in 2,4-DCP concentration with time is illustrated in Fig. 2; in the system without PS, this concentration decreased by only 2.8% in the presence of catalysts over a period of 180 min. In the system with PS alone, approximately 5.3% of the 2,4-DCP was removed from the solution over the same time period. With the addition of 0.2 g L−1 α-MnO2 nanowires to the PS solution, the decrease in the target contaminant (2,4-DCP) accelerated considerably, and the removal efficiency was 90.2% in 180 min. As an activator that is frequently used in PS activation, Fe2+ was added for comparison with the effect of α-MnO2. Given the same molarity, the removal efficiency of 2,4-DCP through the Fe2+-activated PS reaction was only 48.9%. The aforementioned results indicate that reactive free radicals can be efficiently produced with the use of PS activated with α-MnO2 to oxidize 2,4-DCP. Furthermore, the activation efficiency of α-MnO2 was higher than that of Fe2+ given the same molarity in this experimental condition. Recently, Li et al.21 used nano zero valent iron (nZVI) as catalyst to activate PS. Comparing with α-MnO2/PS system studied in the present work, nZVI/PS system needed more catalyst and higher temperature to achieve the best removal efficiency of 2,4-DCP (12.5 Mm PS, 2.0 g L−1 nZVI, 30 mg L−1 DCP, 50 °C). And the removal efficiency was only 34.9% at 30 °C in nZVI/PS system which is much lower than that in α-MnO2/PS system (90.2%) at 30 °C. Sun et al.18 fabricated a new material CuO@Fe3O4 to activate PS to oxidize 2,4-DCP, and 96.9% 2,4-DCP was achieved after 180 min at the optimum condition (10 Mm PS, 0.624 g L−1 CuO@Fe3O4, 100 mg L−1 DCP, 30 °C). Though CuO@Fe3O4 composited the catalytic activity of CuO and Fe3O4, α-MnO2/PS system could attained approximate result at the similar condition (20 Mm PS, 0.2 g L−1 α-MnO2, 100 mg L−1 DCP, 30 °C). These results indicated that α-MnO2 was a promising candidate for degradation of 2,4-DCP in AOPs process.
 |
| | Fig. 2 Degradation of 2,4-dichlorophenol (2,4-DCP) in different systems ([2,4-DCP]0 = 100 mg L−1; [PS]0 = 20 mM; [α-MnO2]0 = 0.2 g L−1 = 0.613 mM; [Fe2+]0 = 0.613 mM, T = 30 °C). | |
Identification of radicals
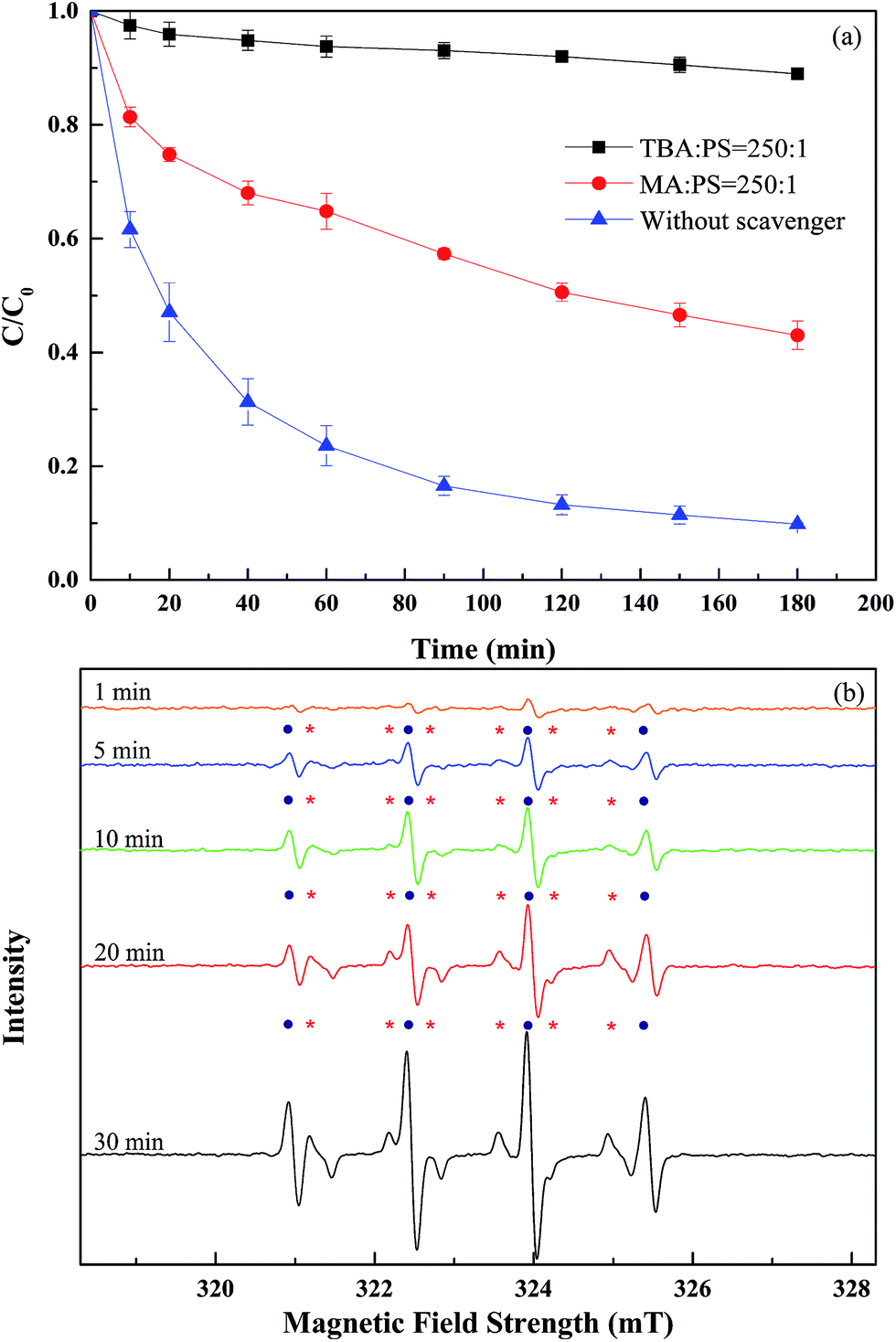
To identify the main reactive species that participate in the PS activation, classical quenching tests were conducted with abundant methyl alcohol (MA) and tert-butyl alcohol (TBA) as quenching agents. The influence of different radical scavengers on the degradation of 2,4-DCP is illustrated in Fig. 3(a). Both MA and TBA can sufficiently quench ˙OH, and the corresponding reaction rates are (1.2–2.8) × 109 and (3.8–7.6) × 108 M−1 s−1.22,23 Given that SO4˙− can be quenched quickly by quenching agents with α-hydrogen, the reaction rates of MA (with α-hydrogen) and TBA (without α-hydrogen) with SO4˙− are (1.6–7.7) × 107 and (4.0–9.1) × 105 M−1 s−1,22–24 respectively. As depicted in Fig. 3(a), the degradation efficiency of 2,4-DCP decreased from 90.2% to either 47.0% or 11.0% with the addition of either excess MA (5 M) or TBA (5 M) over the 180 min reaction period. When either MA or TBA was added into the α-MnO2-activated PS system, the inhibition effect of the latter was considerably stronger than that of the former; this finding is opposite the typical results obtained according to the reaction rates of TBA and MA. A possible reason for this phenomenon may be that a reactive free radical production reaction occurred on the surface of α-MnO2 nanowires under this experimental condition.
 |
| | Fig. 3 (a) Effect of scavengers on 2,4-DCP degradation by PS activated with the α-MnO2 nanowires ([DCP]0 = 100 mg L−1; [PS] = 20 mM; [α-MnO2] = 0.2 g L−1; T = 30 °C); (b) electron parametric resonance spectra of PS and the catalysts in the presence of 0.1 M DMPO (the solid circles represents the ˙OH adduct and the stars denotes the SO4˙− adduct). | |
The dielectric constant is the ratio of the electric displacement in a medium to electric field strength; this ratio can indicate the polarity of the medium. The dielectric constants of TBA and MA are 12.47 and 33.00 at 20 °C;23,25 the polarities of both solvents increase with their respective dielectric constants, as do the water solubility. TBA and MA are hydrophilic compounds; therefore, they may compete with 2,4-DCP in reacting with ˙OH and SO4˙− in the liquid phase instead of accumulating on the surface of the α-MnO2 nanowires. Nonetheless, TBA easily approached the surface of the α-MnO2 nanowires and reacted with the ˙OH and SO4˙− generated in the proximity of the catalyst surface given that the dielectric constant of TBA is smaller than that of MA. Hence, the former easily scavenges reactive free radicals and inhibits the degradation of 2,4-DCP to a larger extent than MA does even if the reaction rate constants of MA with radicals were larger than those of TBA. Therefore, we can speculate that radicals were generated on the catalyst surface in the α-MnO2 nanowires that activate the PS system. Moreover, the reaction between 2,4-DCP and radicals most likely occurred in the boundary layer on the surface of these nanowires. The reactive free radical species mainly participated in the 2,4-DCP oxidation of either ˙OH or SO4˙−.
To further clarify the reactive species in the system, EPR experiments were performed to detect and to comprehensively evaluate the absence of ˙OH and SO4˙−. As shown in Fig. 3(b), characteristic peaks of DMPO–OH adducts (with the special hyperfine coupling constants a(N) = a(H) = 1.49 mT) and characteristic peaks of DMPO–SO4 adducts (with the special hyperfine coupling constants a(N) = 1.38 mT, a(H) = 0.98 mT, a(H) = 0.14 mT, a(H) = 0.08 mT)26,27 were observed and the peak intensity increased with reaction time. The results indicated that both ˙OH and SO4˙− were generated in catalytic reaction and the concentrations of these two radicals increased with the reaction time. However, the intensity of DMPO–SO4 adducts signals was much weaker than that of DMPO–OH adducts signals, which indicating that concentration of SO4˙− was much lower than that of ˙OH. This phenomenon might be due to the fast transformation of DMPO–SO4 adducts to DMPO–OH adducts through nucleophilic substitution.27
Kinetic modeling of 2,4-DCP degradation
A kinetic study was conducted based on the designed experimental conditions. A typical process involving the activation of the PS reaction by α-MnO2 nanowires may be described as a radical chain reaction; ![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Mn(IV)/Mn(III) represents the redox-active manganese surface sites. The one-electron reduction of Mn(IV) facilitates the formation of PS radical (S2O8˙−). In addition, Mn(III) can be oxidized by PS to regenerate Mn(IV) with the production of SO4˙− and sulfate (as expressed in eqn (1) and (2)), which can then initiate radical chain reactions.28 As indicated in eqn (3)–(6), ˙OH and S2O8˙− can be produced via radical chain propagation reactions. The quenching reactions of the radicals between SO4˙− and the ˙OH and 2,4-DCP degradation by these radicals serve as chain termination steps (as expressed in eqn (7)–(11)).
Mn(IV)/Mn(III) represents the redox-active manganese surface sites. The one-electron reduction of Mn(IV) facilitates the formation of PS radical (S2O8˙−). In addition, Mn(III) can be oxidized by PS to regenerate Mn(IV) with the production of SO4˙− and sulfate (as expressed in eqn (1) and (2)), which can then initiate radical chain reactions.28 As indicated in eqn (3)–(6), ˙OH and S2O8˙− can be produced via radical chain propagation reactions. The quenching reactions of the radicals between SO4˙− and the ˙OH and 2,4-DCP degradation by these radicals serve as chain termination steps (as expressed in eqn (7)–(11)).| |
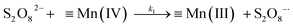 | (1) |
| |
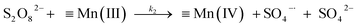 | (2) |
| |
 | (3) |
| |
 | (4) |
| |
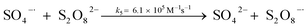 | (5) |
| |
 | (6) |
| |
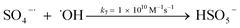 | (7) |
| |
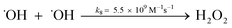 | (8) |
| |
 | (9) |
| |
 | (10) |
| |
 | (11) |
To establish a kinetic model for the degradation reaction, we presume that 2,4-DCP was mainly degraded by SO4˙− and ˙OH. The corresponding kinetic equation for DCP and the radicals can be expressed as follows:
| |
 | (12) |
| |
 | (13) |
| |
 | (14) |
Given that the lifetimes of SO4˙− and ˙OH are merely microseconds long,29 their concentrations are normally considered to be constantly low. Moreover, the corresponding change rates approach zero at a certain time on the basis of the pseudo-steady state assumption. Thus, [SO4˙−] and [˙OH] can be derived as eqn (15) and (16) (derivational process see eqn (S4) to (S11)†):
| |
 | (15) |
| |
 | (16) |
where [
Si] represents the scavengers of SO
4˙
−, such as OH
−, SO
4˙
−, and ˙OH, and [
Sj] represents the scavengers of ˙OH, including S
2O
82−, SO
4˙
−, and ˙OH.
The consumption of Mn(IV) can be expressed by eqn (17).
| |
 | (17) |
As a reaction catalyst, Mn(IV) concentration should be equal to the initial dosage, and the change rate is zero at a certain time. Thus, eqn (17) can be transformed as follows:
| |
 | (18) |
The consumption of S2O82− can be expressed as in eqn (19).
| |
 | (19) |
As per a comparison of eqn (3) and (7), the reaction rate constant of eqn (5), k5, is low; thus, SO4˙− is more likely to react with OH− or ˙OH than with S2O82− and k5[SO4˙−][S2O82−] can be neglected. As with SO4˙−, k6[S2O82−][˙OH] can be disregarded. The concentration of SO4˙− in solution is considerably lower than that of S2O82−; therefore, the k9[SO4˙−][SO4˙−] in eqn (19) can be neglected. Thus, [S2O82−] can be derived from eqn (18) and (19) as follows:
| |
[S2O82−] = [S2O82−]0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) e−2k1[Mn(IV)]t e−2k1[Mn(IV)]t
| (20) |
Substituting eqn (15), (16), (18) and (20) into eqn (12) yields the following:
| |
 | (21) |
To simplify eqn (21), let a = −[k10 + k11k3[OH−]/∑kqr[Sj]]k1[Mn(IV)][S2O82−]0/∑ksr[Si] and b = 2k1[Mn(IV)]. The aforementioned equation can be rearranged in the simplified form shown below.
| |
 | (22) |
The integrated form of eqn (22) is:
| |
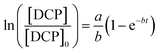 | (23) |
Hence, if the relationship between ln([DCP]/[DCP]0) versus reaction time t matches eqn (23), then the DCP degradation process follows the new reaction dynamic model. The experiments conducted did not only demonstrate this degradation but also verified the proposed models.
Evaluation of the kinetic model in various systems
To verify the kinetic model and to determine the effect of initial conditions on 2,4-DCP degradation, several parameters were investigated, including catalyst dosage, PS concentration, pH, and temperature.
The influence of PS concentration on 2,4-DCP degradation was identified under a fixed α-MnO2 dosage (0.2 g L−1), a temperature of 30 °C, and a PS concentration ranging from 5 mM to 50 mM. The results are depicted in Fig. 4(a). As presented in Fig. 4(a1), 2,4-DCP degradation efficiency increased from 66.1% to 90.2% during the 180 min reaction period when PS concentration increased from 5 mM to 20 mM; however, the removal efficiency changed slightly when PS concentration increased from 20 mM to 50 mM. At low concentrations ranging from 5 mM to 10 mM, the available PS was insufficient to generate reactive free radicals and oxidize the pollutants. As the PS concentration increased to 20 mM, the amount of radicals produced was adequate and the removal efficiency was maximized; nonetheless, a further increase in PS did not facilitate 2,4-DCP degradation, possibly because the number of active sites on the fixed dosage of the catalyst α-MnO2 in the reaction solution was insufficient. In consideration of economy and usability, a concentration of 20 mM was applied in the subsequent experiments. The fitted curves and coefficients of the new kinetic model are illustrated in Fig. 4(a2); the 2,4-DCP degradation curve can fit well with the new kinetic model, and the corresponding regression coefficient values are high (R2 > 0.99).
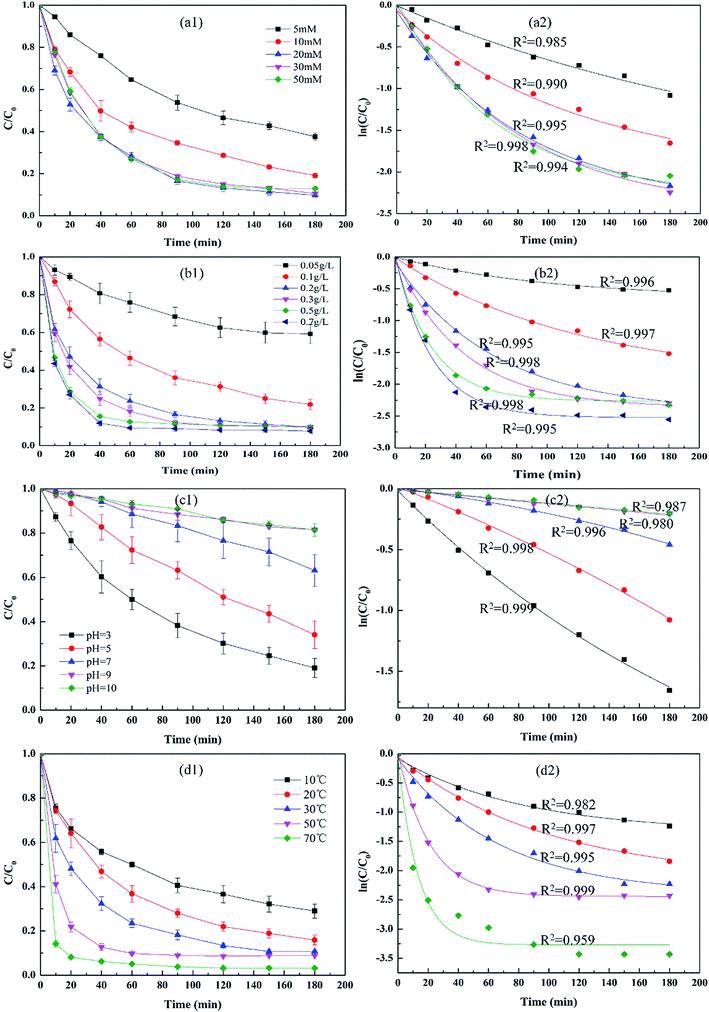 |
| | Fig. 4 Effect of different initial parameters on 2,4-DCP degradation in the activation of PS catalyzed by α-MnO2. (a) PS concentration; (b) catalyst dosage; (c) pH; and (d) temperature. With the exception of the investigated parameter, the remaining parameters were fixed at PS = 20 mM, α-MnO2 = 0.2 g L−1, 2,4-DCP = 100 mg L−1, and temperature = 30 °C. | |
To determine the effect of catalyst dosage on 2,4-DCP degradation, a set of experiments were conducted at 20 mM PS and 30 °C for 180 min. The α-MnO2 nanowires dosage ranged from 0.05 g L−1 to 0.70 g L−1; with this increase, the removal efficiency of 2,4-DCP generally increased from 49.8% to 92.2%. When the α-MnO2 nanowires dosage ranged from 0.05 g L−1 to 0.20 g L−1, 2,4-DCP degradation accelerated significantly from 49.8% to 90.2%. This outcome may be attributed to the increased amount of catalysts in addition to the active sites on the α-MnO2 nanowires, which facilitated the activation of PS to produce reactive free radicals for the oxidation of 2,4-DCP. When the α-MnO2 nanowires dosage increased from 0.2 g L−1 to 0.7 g L−1, the removal efficiency of 2,4-DCP increased only slightly from 90.2% to 92.2%. As per the aforementioned results, a α-MnO2 dosage of 0.2 g L−1 was applied in subsequent experiments. The fitted curves between ln(C/C0) versus reaction time are illustrated in Fig. 4(b2) and conform to the new kinetic model with high regression coefficient values (R2 > 0.995).
pH level is an important factor in the activated PS oxidation system; thus, the influence of initial pH on 2,4-DCP degradation through α-MnO2 activated PS was determined at different pH levels ranging from 3.0 ± 0.2 to 10.0 ± 0.2. The experiments were conducted at a temperature of 30.0 °C, 20.0 mM PS concentration, 0.2 g L−1 of catalyst, and 100.0 mg L−1 2,4-DCP. pH level was previously adjusted with 1 M HCl and 1 M NaOH, and the results are presented in Fig. 4(c1) and (c2). As per Fig. 4(c1), the degradation efficiencies of 2,4-DCP are 80.9%, 66.0%, 36.9%, 18.5%, and 18.4% at different pH levels within 180 min; the removal efficiency was maximized at pH = 3. When pH ranged between 3 and 9, degradation efficiency evidently decreased with an increase in pH and changed slightly when the pH level reached 10. This phenomenon may be attributed to the fact that ˙OH is the predominant radical as a result of the reaction between SO4˙− and OH− (reaction (3)). The standard reduction potentials of ˙OH are 1.8 and 2.7 V in neutral and acidic solutions, respectively. A high pH lowers the oxidizability of ˙OH,22,29 which in turn limits 2,4-DCP oxidation. The shortened lifetime of ˙OH may also weaken 2,4-DCP degradation. The surface charge of the MnO2 catalyst at different pH levels may be another factor; as per previous research, the surface charge of metal oxides is dependent on the relationship between solution pH and the pHpzc of metal oxides.30,31 The surface charge of metal oxides is positive at pH < pHpzc and negative at pH > pHpzc. The pHzpc of several types of manganese oxide has been reported by Prélot et al. and pHzpc of α-MnO2 was 4.5/4.6.32 When solution pH was 3 the surface charge of α-MnO2 was positive. It is easy for S2O82− to contact with the surface of α-MnO2 because of the opposite charge. Under the experimental condition pH > pHzpc (pH = 5–10), the surface charge of the catalyst is negative. As a result of the generation of a repulsive force, the interaction between the negatively charged surface and S2O82− is reduced; in turn, the generation of reactive free radicals is limited.30 This limitation reduces 2,4-DCP removal efficiency. As depicted in Fig. 4(c2), the experimental data fit well with the new kinetic model, with high regression coefficient values (R2 > 0.980) under different pH conditions.
The influence of temperature on the oxidation of 2,4-DCP by α-MnO2-activated persulfate was determined; the initial dosages of PS and α-MnO2 nanowire catalysts were 20 mM and 0.2 g L−1, respectively. Moreover, the selected temperatures ranged between 10 °C and 70 °C. The results are illustrated in Fig. 4(d1) and (d2); according to Fig. 4(d1), 2,4-DCP degradation was strongly enhanced by an increase in temperature. When the temperature increased from 10 °C to 70 °C, the removal efficiency of 2,4-DCP increased from 81.0% to 96.8% in 180 min. A high temperature can also shorten the oxidation reaction time from 180 min to approximately 90 min; the involvement of this phenomenon in the activation process is attributed to the fact that the thermal mobility of the reactant was accelerated at such temperatures; as a result, molecular collision was enhanced. PS can thus be activated efficiently by heat to generate reactive free radicals.33 These radicals are the critical oxidants in 2,4-DCP. The experimental data fitted well in the new kinetic model with high regression coefficient values (R2 > 0.959), as indicated in Fig. 4(d2).
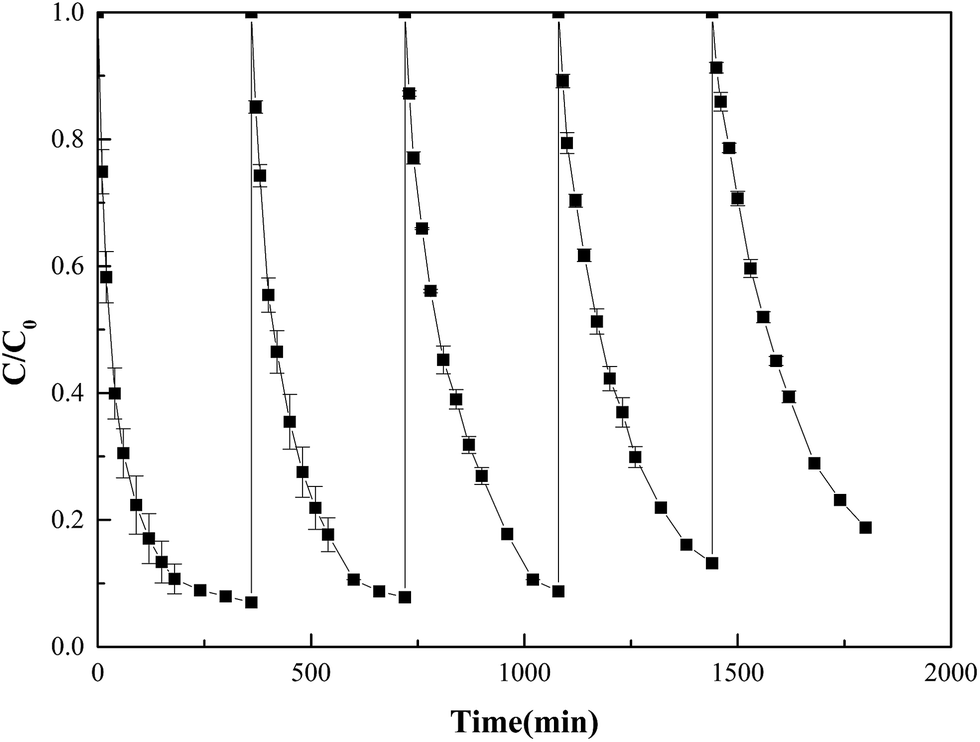
Recycle and reuse of α-MnO2 nanowires
To evaluate the reusability of the catalyst, α-MnO2 nanowires were used repeatedly after washing with water five times to activate PS. As displayed in Fig. 5, 2,4-DCP degradation efficiency declined from 93.0% to 81.2% in each of the 360 min reactions after the catalyst was reused five times. Reactions (1) and (2) suggest that Mn(IV) was reduced by S2O82− to Mn(III) and then oxidized back to Mn(IV) during the activation process. The α-MnO2 nanowires acted as a catalyst rather than as an activator in the oxidation of 2,4-DCP by activated PS. In the activated persulfate solution, the concentration of leached Mn ions was low and could be neglected.34 Catalyst loss may not be the cause of the reduced 2,4-DCP oxidation efficiency. The removal efficiency attained after five reuses reduces activity; this occurrence can be ascribed to the adsorption of reaction intermediates on the α-MnO2 nanowires surface. The adsorbed intermediates cannot be removed simply by washing with water; thus, catalyst activity was inhibited. Nonetheless, the α-MnO2 nanowires catalyst continued to exhibit high activity after five reuses. Thus, these nanowires are a promising catalyst for use in the PS activation process.
 |
| | Fig. 5 Reusability of the α-MnO2 nanowires catalyst. | |
Conclusions
In this study, α-MnO2 nanowires were synthesized through a hydrothermal process and efficiently activated PS for 2,4-DCP oxidation. The composite was characterized by XRD, HRTEM, and EDX. Under the present experiment conditions, a new kinetic model was established by fitting ln(C/C0) and reaction time. During the 180 min reaction period, the degradation efficiency of 2,4-DCP was maximized (90.2%) at an initial PS dosage of 20.0 mM in the presence of 0.2 g L−1 α-MnO2 and under a temperature of 30.0 °C. Furthermore, the removal efficiency of 2,4-DCP increased with PS dosage, catalyst dosage, and temperature as well as with a decrease in the initial pH level. Based on the results of the quenching tests and EPR, both ˙OH and SO4˙− were determined to be the critical reactive free radicals in the α-MnO2-activated PS system. In addition, the α-MnO2 nanowires exhibit good stability and reusability after five reuses in the activation PS system. The result of this study suggests that α-MnO2 nanowires are efficient catalyst for activating PS to remove organic contaminants in wastewater.
Acknowledgements
This study was funded by the Key Project of National Natural Science Foundation of China (No. 41530636), Specific Research of the Environmental Nonprofit Research (No. 2013A073), the National Natural Science Foundation of China (No. 41302184), and National Natural Science Foundation of China (No. 41471252), as well as interdisciplinary research project of Jilin University.
Notes and references
- Z. Sun, X. Wei, Y. Han, S. Tong and X. Hu, J. Hazard. Mater., 2013, 244, 287–294 CrossRef PubMed.
- F. Shaarani and B. Hameed, Desalination, 2010, 255, 159–164 CrossRef CAS.
- T. Zhou, Y. Li, F.-S. Wong and X. Lu, Ultrason. Sonochem., 2008, 15, 782–790 CrossRef CAS PubMed.
- L. F. Liu, P. H. Zhang and F. L. Yang, Sep. Purif. Technol., 2010, 70, 354–361 CrossRef CAS.
- Y. Wang, H. Ren, H. Pan, J. Liu and L. Zhang, J. Hazard. Mater., 2015, 286, 269–275 CrossRef CAS PubMed.
- M. A. Talano, D. C. Busso, C. E. Paisio, P. S. González, S. A. Purro, M. I. Medina and E. Agostini, Environ. Sci. Pollut. Res., 2012, 19, 2202–2211 CrossRef CAS PubMed.
- M. A. Talano, S. Frontera, P. González, M. I. Medina and E. Agostini, J. Hazard. Mater., 2010, 176, 784–791 CrossRef CAS PubMed.
- C. Liang and I. L. Lee, J. Contam. Hydrol., 2008, 100, 91–100 CrossRef CAS PubMed.
- J. Yan, M. Lei, L. Zhu, M. N. Anjum, J. Zou and H. Tang, J. Hazard. Mater., 2011, 186, 1398–1404 CrossRef CAS PubMed.
- Y. S. Zhao, C. Sun, J. Q. Sun and R. Zhou, Sep. Purif. Technol., 2015, 142, 182–188 CrossRef CAS.
- H. Zhao, H.-J. Cui and M.-L. Fu, RSC Adv., 2014, 4, 39472–39475 RSC.
- E. Saputra, S. Muhammad, H. Sun, H.-M. Ang, M. O. Tadé and S. Wang, J. Colloid Interface Sci., 2013, 407, 467–473 CrossRef CAS PubMed.
- C. Yu, G. Li, L. Wei, Q. Fan, Q. Shu and J. C. Yu, Catal. Today, 2014, 224, 154–162 CrossRef CAS.
- W. Zhang, Z. Yang, X. Wang, Y. Zhang, X. Wen and S. Yang, Catal. Commun., 2006, 7, 408–412 CrossRef CAS.
- L. Xu, C. Xu, M. Zhao, Y. Qiu and G. D. Sheng, Water Res., 2008, 42, 5038–5044 CrossRef CAS PubMed.
- E. Saputra, S. Muhammad, H. Sun, H. M. Ang, M. Tade and S. Wang, Environ. Sci. Technol., 2013, 47, 5882–5887 CrossRef CAS PubMed.
- L. Hou, H. Zhang and X. Xue, Sep. Purif. Technol., 2012, 84, 147–152 CrossRef CAS.
- C. Sun, R. Zhou, E. Jianan, J. Sun and H. Ren, RSC Adv., 2015, 5, 57058–57066 RSC.
- E. Saputra, S. Muhammad, H. Sun, A. Patel, P. Shukla, Z. Zhu and S. Wang, Catal. Commun., 2012, 26, 144–148 CrossRef CAS.
- A. M. Hashem, H. M. Abuzeid, A. M. Abdel-Latif, H. M. Abbas, H. Ehrenberg, S. Indris, A. Mauger, H. Groult and C. M. Julien, ECS Trans., 2013, 50, 125–130 CrossRef.
- R. Li, X. Jin, M. Megharaj, R. Naidu and Z. Chen, Chem. Eng. J., 2015, 264, 587–594 CrossRef CAS.
- G. V. Buxton, C. L. Greenstock, W. P. Helman and A. B. Ross, J. Phys. Chem. Ref. Data, 1988, 17, 531–886 CrossRef.
- H.-Y. Liang, Y.-Q. Zhang, S.-B. Huang and I. Hussain, Chem. Eng. J., 2013, 218, 384–391 CrossRef CAS.
- P. Neta, R. E. Huie and A. B. Ross, J. Phys. Chem. Ref. Data, 1988, 17, 1027–1284 CrossRef CAS.
- D. Lide and W. Haynes, CRC handbook of chemistry and physics: a ready-reference book of chemical and physical data, ed. David R. Lide and WM“Mickey” Haunes, CRC, Boca Raton, Fla, 2009 Search PubMed.
- Y. Wang, H. Sun, X. Duan, H. M. Ang, M. O. Tadé and S. Wang, Appl. Catal., B, 2015, 172, 73–81 CrossRef.
- J. Zou, J. Ma, L. Chen, X. Li, Y. Guan, P. Xie and C. Pan, Environ. Sci. Technol., 2013, 47, 11685–11691 CrossRef CAS PubMed.
- H. Liu, T. A. Bruton, F. M. Doyle and D. L. Sedlak, Environ. Sci. Technol., 2014, 48, 10330–10336 CrossRef CAS PubMed.
- S. Wang, N. Zhou, S. Wu, Q. Zhang and Z. Yang, Ultrason. Sonochem., 2015, 23, 128–134 CrossRef CAS PubMed.
- J. Liu, Z. Zhao, P. Shao and F. Cui, Chem. Eng. J., 2015, 262, 854–861 CrossRef CAS.
- Y. Xin Zhang, X. Long Guo, M. Huang, X. Dong Hao, Y. Yuan and C. Hua, J. Phys. Chem. Solids, 2015, 83, 40–46 CrossRef.
- B. Prélot, C. Poinsignon, F. Thomas, E. Schouller and F. Villiéras, J. Colloid Interface Sci., 2003, 257, 77–84 CrossRef.
- C. Liang and C. J. Bruell, Ind. Eng. Chem. Res., 2008, 47, 2912–2918 CrossRef CAS.
- Y. Wang, H. Sun, H. M. Ang, M. O. Tadé and S. Wang, Appl. Catal., B, 2015, 164, 159–167 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Text S1. See DOI: 10.1039/c6ra00008h |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.