
Effective saccharification of holocellulose over multifunctional sulfonated char with fused ring structures under microwave irradiation
Abstract
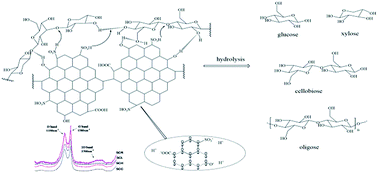
An environmentally benign process for hydrolysis of holocellulose conversion into sugars has been presented in this work, using sulfonated char (SC) catalysts derived from microcrystalline cellulose, hemicellulose, lignin and lignin-rich residues. Significantly, the SC catalysts exhibited remarkable hydrolysis performance for holocellulose under microwave irradiation, especially with sulfonated char from lignin (SCL) and sulfonated char from residues (SCR). The maximum conversion of holocellulose could reach 82.9% using SCR catalyst, with 26.8 wt% content of monose and 72.2 wt% content of oligose. The as-synthesized SC catalysts were characterized using scanning electron microscopy (SEM), X-ray diffraction (XRD), Raman spectroscopy (RS), Fourier transform infrared spectroscopy (FT-IR), thermal gravity analysis (TG) and elemental analysis (EA), which demonstrated that the SC catalysts possess a graphene-like fused ring structures, bearing SO3H, COOH and phenolic OH groups. The excellent catalytic performance could be attributed to the synergetic combination of the fused ring structures and multifunctional groups in the SC catalysts. This process could offer a promising strategy for efficient conversion of biomass in the future.
 Please wait while we load your content...
Please wait while we load your content...

