
Quantitative description of the SABRE process: rigorous consideration of spin dynamics and chemical exchange†
Abstract
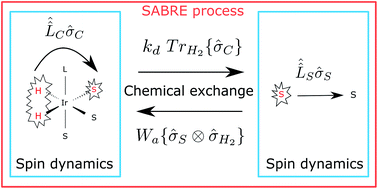
A consistent theoretical description of the spin dynamics and chemical kinetics underlying the SABRE (Signal Amplification By Reversible Exchange) process is proposed and validated experimentally. SABRE is a promising method for Nuclear Magnetic Resonance (NMR) signal enhancement, which exploits the transfer of strong non-thermal spin order from parahydrogen (the H2 molecule in its singlet spin state) to a substrate in a transient organometallic complex. A great advantage of the SABRE method is that the substrate acquires strong nuclear spin polarization without being modified chemically, as it is only transiently bound to the complex. However, for the same reason theoretical treatment of SABRE meets difficulties because of the interplay of the spin dynamics with the association–dissociation reactions of the SABRE complex. Here we propose a quantitative model, which takes into account both the spin evolution in the SABRE complex and the substrate exchange between the free and bound forms. The model allows for the calculation of the substrate spin polarization dependency on various parameters, such as the external magnetic field strength and complex association–dissocation rates, and enables the simulation of experimental data for the SABRE time dependence. This investigation opens new insights into the SABRE process and can be generalized to treat more complex cases, such as SABRE facilitated by NMR pulses.
 Please wait while we load your content...
Please wait while we load your content...

