
Role of the non-conserved amino acid asparagine 285 in the glycone-binding pocket of Neosartorya fischeri β-glucosidase†
Abstract
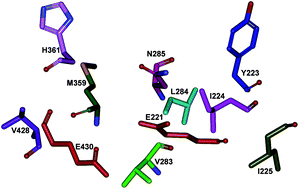
Neosartorya fischeri β-glucosidase (NfBGL595) is distinguished from other BGLs by its high turnover for p-nitrophenyl β-D-glucopyranoside (pNPG) and flavones. The role of non-conserved amino acids in the glycone-binding pocket of NfBGL595 was studied using sequence alignment and homology modeling, followed by site-directed mutagenesis. Nine amino acids (Y223, I224, I225, V283, L284, N285, M359, H361, and V428) were identified as variable residues around the active site residues, E221 and E430, and selected for mutagenesis. Mutation of the residues to Ala resulted in a drastic alteration in the kcat and Km values when compared to the wild type NfBGL595. Among these nine residues, mutation of N285 to Ala resulted in a complete loss of activity toward pNPG and flavonoid glucosides. Further mutation, structural, and docking analyses revealed that residue N285 is crucial in maintaining the pKa and polarity around E221, which is surrounded by non-polar residues. This study suggests the importance of the pKa and microenvironment around the active site pocket for BGL catalysis.
 Please wait while we load your content...
Please wait while we load your content...

