
A pH-driven molecular shuttle based on rotaxane-bridged periodic mesoporous organosilicas with responsive release of guests†
Abstract
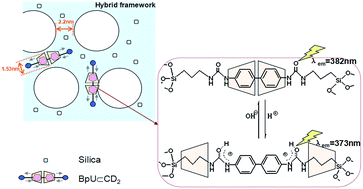
As a strategy for achieving integration of two-state rotaxane based molecular switches and ordered solid-state frameworks, a pH-driven molecular shuttle was immobilized into the framework of the periodic mesoporous organosilicas (PMOs) that possessed enough free space to accommodate the mechanical motion of β-cyclodextrins (β-CDs). In this molecular shuttle, β-CDs threaded a symmetrical molecular thread composed of a biphenyl unit, two ureido and propyl groups, and were end-trapped mechanically by two siloxane stoppers. The β-CDs, as the shuttles, could be reversibly translocated along the thread by the pH stimuli in the rigid framework, accompanied with the change of fluorescence emission of the biphenyl units. Particularly, the PMOs could be employed as a pH-controllable smart-release platform via the reciprocating movement properties of the molecular shuttle, and accelerated cargo release was achieved after acidification. Furthermore, the PMO materials show very low cytotoxicity and fine biocompatibility, which ensure their potential in biomedical applications.
 Please wait while we load your content...
Please wait while we load your content...

