
Ni-based chromite spinel for high-performance supercapacitors†
Abstract
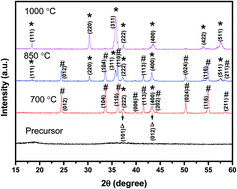
NiCr2O4 has been prepared through the easy precipitation reaction of Ni2+ and Cr3+ (1 : 2 mol ratio) in aqueous NH3 followed by annealing at a high temperature. This is the first time that NiCr2O4 has been used as an electrode material for a supercapacitor. As expected, the NiCr2O4 exhibits good supercapacitive performance: at a current density of 0.6 A g−1, the specific capacitances of NiCr2O4 in the three-electrode and two-electrode setups are 422 and 187 F g−1, respectively; the energy density for the NiCr2O4 symmetric device reaches 6.5 W h kg−1 at a power density of 3000 W kg−1; and about 80% of the capacitance is retained after 2000 charge/discharge cycles. Therefore, research on NiCr2O4 as a supercapacitor material is necessary.
 Please wait while we load your content...
Please wait while we load your content...

