
Untargeted metabolomic analysis using LC-TOF/MS and LC-MS/MS for revealing metabolic alterations linked to alcohol-induced hepatic steatosis in rat serum and plasma†
Huan Wuab and
Fang Feng*abc
aDepartment of Pharmaceutical Analysis, China Pharmaceutical University, Nanjing 210009, China. E-mail: fengfang1@hotmail.com; Tel: +86 025 83271301
bKey Laboratory of Drug Quality Control and Pharmacovigilance (Ministry of Education), China Pharmaceutical University, Nanjing 210009, China
cState Key Laboratory of Natural Medicine, China Pharmaceutical University, Nanjing 210009, China
First published on 7th March 2016
Abstract
Alcohol-induced hepatic steatosis (AHS), an early stage of alcoholic liver disease, is characterized by large amounts of fat deposited in hepatocytes. Although some important biomarkers have been identified, there still is no systematic view of AHS-associated biomarker alteration in vivo. Blood matrix choice is a fundamental consideration for biomarker exploration. In the present study, a reliable untargeted metabolomic method based LC-TOF/MS and LC-MS/MS was developed to (i) interrogate overall differences of endogenous metabolites in rat serum and plasma, and investigate which of the two matrices is more suitable for the discovery of endogenous biomarkers, (ii) analyze metabolic alterations linked to incident AHS. As a result, metabolite level differences between serum and plasma in normal rats were mainly related to 7 peptides, 8 lysophosphatidylcholines, 3 phosphatidylcholines, 2 heterocyclic compounds, 7 polyunsaturated fatty acids and their oxidative fatty acids. Notably, 7 characteristic peptides were observed only in serum. All the altered metabolites were associated with clotting cascade reaction, indicating that serum conceals the changes in metabolome that have occurred in vitro. The results demonstrated that plasma which represented the original properties of the blood sample was a more suitable matrix to explore potential biomarkers. In the plasma untargeted metabolomic study, the metabolic alterations linked to AHS were mainly involved in phospholipid metabolism, amino acids metabolism, fatty acid metabolism, cholesterol metabolism and sphingolipid metabolism. These findings provided a systematic view of metabolic alterations linked to AHS, demonstrating the untargeted metabonomic method was a robust method for examining the molecular mechanisms of disease.
1. Introduction
Untargeted metabolomics has emerged as a powerful method for the discovery of disease biomarkers, providing fundamental insights into cellular biochemistry and clues related to disease pathogenesis over the last two decades.1,2 It consists of the global profiling of low-molecular-weight metabolites in organisms and identifying the most meaningful metabolites which play important roles in biological pathways. Mass spectrometry (MS)3,4 and nuclear magnetic resonance (NMR) spectroscopy5,6 are two principal analytical platforms used to perform untargeted metabolomics in recent years. Although NMR provides conclusive structural information and high reproducible data about metabolites in vivo, it suffers from limitations in sensitivity and chemical resolution.7 In contrast, MS provides less-conclusive structural information, but given its high sensitivity, good specificity and large dynamic range, it allows for the acquisition of more data from each biological sample.8 Because of their full-spectrum detection capabilities and high mass accuracy, high-resolution mass spectrometers such as time-of-flight (TOF), Orbitrap, or hybrids mass analyzers such as quadrupole-TOF (QTOF) and Q Orbitrap are adequate to detect many more chemical species and develop untargeted metabolomic approaches in complex biological matrixes.9,10Alcoholic liver disease (ALD) is a chronic disorder characterized by excessive ingestion of alcohol and manifested as a broad spectrum of disorders, ranging from hepatic steatosis, steatohepatitis, fibrosis and cirrhosis (Laennec’s cirrhosis).11 The earliest liver injury observed in alcoholics is steatosis which is characterized by large amounts of fat deposited in hepatocytes.12 Alcohol-induced hepatic steatosis (AHS) is largely asymptomatic and the liver damage is reversible after cessation of alcohol consumption, otherwise it advances to irreversible stages of ALD which associated with increased risk of liver cancer.13 Detection of ALD at the stage (AHS) is, therefore, key to ameliorate quality of life, improve therapeutic benefit and reduce healthcare burden.
Although some important biomarkers have been identified,12,14 there still lacks a systematic view of AHS-associated biological processes in vivo, such as broad metabolite level, which is pivotal to clarify the physiological mechanism of disease. Matrix choice is a fundamental consideration for biomarker exploration. Peripheral blood possesses a wealth of information and has been shown to establish the overall pathophysiology mapping of an individual.15 It does satisfy the criteria of minimal invasiveness and reasonable cost compared with the collection of tissue samples by biopsy.16 Therefore, serum and plasma samples are widely applied in metabolomic analysis of mammalian systems.17 However, few studies have investigated potential differences in serum and plasma metabolomes and explored which matrix could capture optimal metabolite information coverage for disease.
In this study, we aimed to (i) interrogate serum and plasma in order to identify overall differences in the metabolite levels, and investigate which of the two matrixes is more suitable for the discovery of endogenous biomarkers, (ii) analyze metabolic alterations linked to AHS. Toward these aims, an untargeted metabolomic approach based LC-TOF/MS and LC-MS/MS was used to globally profile and identify the low-molecular-weight endogenous metabolites in serum and plasma of normal and AHS rats, respectively. Both univariate statistics and multivariate data analysis (MVDA) were employed to test the analytical reproducibility in terms of detected ion intensities and rapidly screen the interesting metabolites associated with AHS.
2. Experimental
2.1 Chemicals and reagents
Chromatographic-grade methanol and acetic acid were purchased from Merck (Darmstadt, Germany) and Sigma Chemical (St. Louis, USA), respectively. Ultra high purity water was produced by Millipore-Q water purification system (Bedford, USA).2.2 Animals and treatment
All protocols and care of the rats were in accordance with the Guidelines for the Care and Use of Laboratory Animals and approved by the Animal Ethics Committee of China Pharmaceutical University. Male Sprague-Dawley rats (12–14 weeks) weighing 180–220 g were acclimated at 50 ± 20% humidity and 20 ± 2 °C with a 12 h light/dark cycle in an animal breeding room. Purified water and standard chow were provided ad libitum. After 1 week of acclimatization, the rats were randomly divided into the AHS group and control group (n = 8 for each group). Animals were housed individually in cages and provided with the regular Lieber–DeCarli alcohol or pair-feeding (ethanol was substituted isocalorically with carbohydrate) liquid diet for eight weeks in AHS group and control group, respectively. The amount of ethanol (%, w/v) in the liquid diet was 5.00%, 5.14%, 5.29% and 5.43% for every two weeks, respectively. To achieve equal daily energy intake, the rats in the AHS group were fed ad libitum, while control rats were pair-fed the same amount consumed by model rats during the prior day.182.3 Sample collection
All rats were fasted over night before the study. The rats were anesthetized by intraperitoneal injection of urethane (1.6 g kg−1 body weight), and blood samples were collected from hepatic portal vein into heparin anticoagulation (plasma) and no anticoagulant (serum) tubes. The heparin anticoagulation tubes were gently inverted three times to ensure proper mixing of blood with the heparin anticoagulant. The plasma samples were immediately isolated by centrifugation (2500 × g, 10 min, 4 °C) after sampling. The serum samples were kept at 4 °C for 1 h to clot after blood collection, and then centrifuged (2500 × g, 10 min, 4 °C). Volumes of 200 μL plasma or serum samples were aliquoted separately in 2.0 mL microcentrifuge tubes. A quality control pooled (QCP) sample was prepared by mixing aliquots of 20 μL of each of the experimental samples. All samples were stored at −80 °C until sample preparation was initiated.2.4 Analytical design
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 × g was performed for 10 min at 4 °C. A 600 μL aliquot of supernatant was transferred into a fresh microcentrifuge tube and speed-vacuum-dried at room temperature, then reconstituted in 200 μL of 20:80 methanol/water followed by centrifugation (12500 × g, 10 min, 4 °C). A 20 μL aliquot was injected for LC-MS analysis.
500 × g was performed for 10 min at 4 °C. A 600 μL aliquot of supernatant was transferred into a fresh microcentrifuge tube and speed-vacuum-dried at room temperature, then reconstituted in 200 μL of 20:80 methanol/water followed by centrifugation (12500 × g, 10 min, 4 °C). A 20 μL aliquot was injected for LC-MS analysis.The optimized conditions of TOF/MS were: capillary voltage, 4.0 kV/−3.5 kV; drying gas (N2) temperature, 350 °C; drying gas (N2) flow rate, 12.0 L min−1; Oct RFV, 750 V; skimmer voltage, 65 V; fragmentor voltage, 175 V; nebulizer pressure, 35 psi; scan range, 100–1200 Da. To ensure accuracy, the mass-to-charge ratio (m/z) of all ions in the mass spectra were real-time corrected by reference ions (m/z 121.050873 (protonated purine) and 922.009798 (protonated hexakis, (1H,1H,3H-tetrafluoropropoxy)phosphazine (HP-921)) for positive mode, m/z 112.985587 (proton-abstracted ammonium trifluoroacetate (TFA) anion) and 1033.988109 (TFA adduct of HP-0921) for negative mode). The acquisition and analysis of data were controlled by Mass Hunter B.04.00 software.
Each of the samples was analyzed by LC-TOF/MS in both positive and negative ionization mode to obtain metabolite profiles and the analysis order of all test samples was randomized.19 In order to condition the LC-MS system, the QCP sample was injected five times before initiating the run. The QCP sample was re-injected once at the beginning, every four sample injections, and at the end of the run (total of nine injections) to provide a set of data to monitor instrument stability and evaluate reproducibility.
2.5 Data analysis
The S-plots generated by OPLS-DA were used to visualize the relative importance of the different sources of variations and obtain a list of candidates. Variables at VIP (variable importance in the projection) >1 were considered. Features at VIP > 1 with a large jack-knifed confidence interval were excluded19 and the other values were further subjected to unpaired parametric t-test (Welch’s t-test, assuming unequal variance) to determine the significance of each feature. Only tR–m/z pairs with VIP > 1 and p < 0.05 were selected as potential chemical markers.
3. Results and discussion
3.1 Technical reproducibility of the analytical approach
In order to reduce ion suppression induced by co-eluting metabolites of the same class, different mobile phase compositions were screened to obtain LC chromatograms with better peak shape and separation. It was found that 0.1% acetic acid–methanol was the most suitable eluting solvent system. The analytical quality in metabolomics study is of utmost importance as a guarantor for reliable results and accordingly for the success of the entire project. To ensure the robustness of analytical instrumentation and the reproducibility of data, the periodic analysis of QCP samples in the same batch was adopted as a quality assurance strategy in metabolic profiling. The QCP sample is applied for two reasons. The first reason is that the metabolic and sample matrix compositions of QCP samples are closest to the biological samples under study. The second reason is to provide data assessing technical reproducibility of the analytical approach.4Using the 9 QCP samples interspersed throughout the run, both univariate statistics and MVDA were employed to test the analytical reproducibility in terms of detected ion intensities: (1) by calculating the RSD% of detected ion intensities in the QCP samples as a univariate way28 and (2) using PCA as a MVDA method to assess reproducibility. After data extraction, the number of features in positive and negative mode was 537 and 674 for the QCP sample, respectively. After excluding features with 80%-rule29 and %RSD higher than 60%, more than 75% of QCP sample features in positive mode and 71% of QCP sample features in negative mode with %RSD less than 30% were subjected to MVDA (Fig. S1†). The PCA demonstrated a tight grouping of the QCP samples and provided pattern distinction for normal rat plasma, normal rat serum, AHS rat plasma and AHS rat serum samples in positive (Fig. 1A) as well as negative ionization modes (Fig. 1C), implying their high analytical reproducibility across the run and confirming that the tR–m/z pairs observed between the samples were nonsystematic, but biologically related. To further inspect data quality and run order effect on the test samples and QCP samples in positive (Fig. 1A) and negative-ion modes (Fig. 1C), corresponding Hotelling’s T2Range plot (denotes 95 and 99% significance limits, values larger than the yellow limit are suspect (0.05 level), and values larger than the red limit (0.01 level) can be considered serious outliers) was used to catch outliers in data and observe effects of run order in Fig. 1B and D. It is explicit that there was no outlier and no time-related effect on the detected ions. The high reproducibility of the data demonstrated that the quality assurance procedures can deliver the reliability required by an untargeted metabolomic study.
 | ||
| Fig. 1 (A) PCA score plot of all analyzed samples in positive-ion mode with the statistical parameters (R2X = 0.601, Q2 = 0.768), and (B) corresponding Hotelling’s T2Range plot showing sample type to detect any trends in QCP samples and test samples. (C) PCA score plot of all analyzed samples in negative-ion mode with the statistical parameters (R2X = 0.722, Q2 = 0.567), and (D) corresponding Hotelling’s T2Range plot. Normal rat plasma (green dots), normal rat serum (blue squares), AHS rat plasma (brown triangles) and AHS rat serum (yellow inverted triangles) samples as well as QCP samples (turquoise diamonds) are shown. | ||
3.2 Differences in metabolite profiles between serum and plasma samples
The differences between the two specimens have been studied by Roche Modular P analyzer and Vitros 950 analyzer on a biochemical level30 and have been analyzed for proteomics or metabolomics applications by LC-MS,31 NMR32 and GC-MS.33 To better investigate and visualize the origin of specimen type differences, both Welch’s t-test and MVDA (PCA and OPLS-DA) were established as described below. After data extraction and quality assurance procedures, supervised OPLS-DA was employed to differentiate the tR–m/z pairs between the heparin plasma and serum samples. In the score plots of OPLS-DA, it was found that heparin plasma samples (green dots) and serum samples (blue squares) cluster into two discrete groups (Fig. 2A and C), implying that potential discriminant tR–m/z pairs contributed to the good separation. The discriminant tR–m/z pairs were visualized and filtered by the S-plot and VIP value from OPLS-DA, respectively. As seen from Fig. 2B and D, the blue 4-point star graph is the S-plot, where the further the distance of the tR–m/z pairs from the origin point, the higher the confidence level of the tR–m/z pairs contributed to the clustering observed in the score plots of OPLS-DA. Furthermore, the tR–m/z pairs with VIP > 1.0 selected from S-plot were further subjected to Welch’s t-test to determine the significance of each variable. Finally, 16 and 15 interesting tR–m/z pairs (in each respective ion mode) were extracted and their tentative assignments in the online available databases are shown in Table S1.† The metabolites included 7 peptides, 8 lysophosphatidylcholines (lysoPCs), 3 phosphatidylcholines (PCs), 2 heterocyclic compounds, 7 polyunsaturated fatty acids (PUFAs) and their oxidative fatty acids (oxFAs). Among these, phe–phe, lysoPC(18:1), lysoPC(16:0) and eicosapentaenoic acid were found in both positive- and negative-ion modes.
 | ||
| Fig. 2 Score plots of OPLS-DA in positive-ion mode (A) with the statistical parameters (R2X = 0.645, R2Y = 0.998, Q2 = 0.977, p[CV-ANOVA] = 7.3 × 10−7) and in negative-ion mode (C) with the statistical parameters (R2X = 0.723, R2Y = 0.991, Q2 = 0.961, p[CV-ANOVA] = 7.9 × 10−6) between the serum (blue squares) and heparin plasma (green dots). The corresponding S-plot from OPLS-DA model of the two groups in positive-ion mode (B) and in negative-ion mode (D). | ||
Serum was characterized by the presence of some dominating singly and doubly charged peptides. From the trend plots of S-plot, the tR–m/z pairs of peptides that only existed in the serum constitute the major difference between serum and heparin plasma samples. Taking the tR–m/z 9.77–120.0811 (a singly charged peptide) and tR–m/z 31.21–453.1721 (a doubly charged peptide) in positive-ion dataset for example, the interesting tR–m/z pairs were extracted easily at the top of the S-plot (Fig. 3A and D). Then, ±20 ppm narrow mass window extracted ion chromatograms (EICs) (Fig. 3B and E) and corresponding in-source fragmentations (Fig. 3C and F) were used to further confirm. The characteristic peptides which are only present in serum and not detectable in plasma are believed to emerge from peptides derived from cellular components or the clot, and the release of platelet-derived peptides during the coagulation process. This ex vivo generation of multiple peptides have been speculated to potentially affect proteome profiling31 and could also lead to a potential misinterpretation of serum metabolomics data.
 | ||
| Fig. 3 Intensity trend plots, EICs and corresponding in-source fragmentations of a singly charged peptide (tR–m/z 9.77–120.0811, fragmentation of phenylalanine–phenylalanine) (A–C) and a doubly charged peptide (tR–m/z 31.21–453.1721) (D–F) in positive-ion mode (X-axis represents serum (blue squares) and heparin plasma (green dots) samples and Y-axis represents response in (A) and (D)). | ||
8 lysoPCs(lysoPC(15:0), lysoPC(16:0), lysoPC(16:1), lysoPC(17:0), lysoPC(18:0), lysoPC(18:1), lysoPC(18:2), lysoPC(20:4)), 3 PCs (PC(34:2), PC(36:4), PC(40:5)) and 5 PUFAs and their oxFAs (eicosapentaenoic acid, arachidonic acid, 12-hydroxyheptadecatrienoic acid, 12-hydroxyeicosapentaenoic acid and 5,6-dihydroxyeicosatrienoic acid) were markedly higher in serum than in heparin plasma. PCs are a class of phosphoglycerolipids (PLs) that incorporate choline as a headgroup. They are the major components of biomembranes. LysoPCs, as a major bioactive phospholipid component of oxidized low-density lipoprotein, are formed by hydrolysis of sn-2 fatty acyl bond of phospholipids by phospholipase A2 (PLA2).34 The specimen-associated differences among the levels of 8 lysoPCs, 3 PCs, 5 free PUFAs and their oxFAs were also related to the clotting process in vitro. The coagulation process results in thrombin-stimulated platelets releasing PLs, lyso-PLs and PUFAs from biomembrane by activated PLA2 via inositol 1,4,5-phosphate signaling.16 Whereas two oxFAs metabolites (8-hydroxyeicosatetraenoic acid, 5,15-dihydroxyeicosatetraenoic acid) were significantly lower in serum than in heparin plasma. Degradation of the two lipid metabolites in serum may occur during the coagulation process. Another difference between serum and heparin plasma was related to two heterocyclic compounds (hypoxanthine and its metabolite xanthine). The higher level of hypoxanthine and xanthine in serum had been attributed to presence of platelets and erythrocytes, which could continue to release these oxypurines after the blood specimen was drawn.35 Therefore, the levels of these metabolites associated with clotting cascade reaction in serum are unable to reflect real biological processes in vivo.
3.3 Differences in metabolite profiles between normal and AHS rat samples
After data extraction and quality assurance procedures, MVDA was applied to generate a comparison of normal rat serum and AHS rat serum samples, normal rat plasma and AHS rat plasma samples. Score plots of OPLS-DA and the corresponding S-plot between the normal rat serum and AHS rat serum samples are shown in Fig. S2,† the normal rat plasma and AHS rat plasma samples are shown in Fig. S3.† Based on VIP >1.0 of OPLS-DA and p < 0.05 of Welch’s t-test, 35 differential metabolites between the normal rat serum and AHS rat serum, 34 differential metabolites between the normal rat plasma and AHS rat plasma were identified (Table 1). The typical total ion chromatograms of normal rat plasma and AHS rat plasma in positive-ion mode (A, B) and in negative-ion mode (C, D) are shown in Fig. S4.† Metabolite identification was conducted with accurate mass and fragmentation behavior, as well as online database analyses. To illustrate the identification of metabolites, we took the tR–m/z 50.63–524.3713 in positive ion dataset and tR–m/z 55.71–363.2583 in negative ion dataset for example (Fig. 4). In positive ion ESI-MS, two ions at m/z 524.3713 and 546.3529 were extracted at 50.63 min (Fig. 4A). We inferred that the quasi-molecular ions were 524.3713 [M + H]+ and 546.3529 [M + Na]+, respectively. The molecular formula of the metabolite was established as C26H54NO7P. According to the HMDB database and mass fragments supplemented by MS/MS scan (Fig. 4B), the differential metabolite was tentatively assigned as lysoPC(18:0). In negative ion ESI-MS, two ions at m/z 363.2553 [M + CH3COO]− and 339.2029 [M + Cl]− were found at 55.71 min (Fig. 4C) and molecular formula of the metabolite was established as C20H32O2. The fragment ions at 259.24 [M − H − CO2]−, 205.20 [M − H − CO2 − C4H6]− and 163.14 [M − H − CO2 − C4H6 − C3H6]− were produced using target ESI-MS/MS spectra (Fig. 4D). According to the accurate mass and its fragment information, the metabolite was tentatively identified as arachidonic acid.
| Features (tR–m/z) | Adduct | Formula | MW/Da | Diff./ppm | Metabolite | Related pathway | VIP | Intensity response |
|---|---|---|---|---|---|---|---|---|
| a Abbreviations: MW, monoisotopic weight; VIP, variable importance on projection; LysoPC, lysophosphatidylcholine; PG, prostaglandin; Phe, phenylalanine; GCDCA, glycochenodeoxycholic acid; PC, phosphatidylcholine; GDCA, glycodeoxycholic acid. *: response compared to control group samples. | ||||||||
| Positive mode | ||||||||
| 50.63–524.3713 | [M + H]+ | C26H55NO7P | 524.3716 | −0.6 | LysoPC(18:0) |
Phospholipid metabolism | 2.01 | ↓* |
| 3.21–138.0541 | [M + Na]+ | C5H9NO2Na | 138.0531 | 7.2 | L-Proline | Amino acids metabolism | 1.94 | ↑* |
| 55.62–381.2635 | [M + H]+ | C22H37O5 | 381.2641 | −1.6 | 20-Ethyl-PGE2 | Fatty acid metabolism | 1.88 | ↓* |
| 6.39–165.0560 | [M + H]+ | C9H9O3 | 165.0552 | 4.8 | Phenylpyruvic acid | Amino acids metabolism | 1.75 | ↑* |
| 45.18–522.3556 | [M + H]+ | C26H53NO7P | 522.3560 | −0.8 | LysoPC(18:1) |
Phospholipid metabolism | 1.73 | ↓* |
| 49.99–351.2521 | [M + H]+ | C21H35O4 | 351.2535 | −4.0 | PGA1 methyl ester | Fatty acid metabolism | 1.62 | ↑* |
| 50.57–546.3534 | [M + H]+ | C28H53NO7P | 546.3560 | −4.8 | LysoPC(20:3) |
Phospholipid metabolism | 1.49 | ↑* |
| 3.11–132.1019 | [M + H]+ | C6H14NO2 | 132.1025 | −4.5 | L-Leucine | Amino acids metabolism | 1.47 | ↑* |
| 43.71–518.3235 | [M + Na]+ | C24H50NO7PNa | 518.3223 | 2.3 | LysoPC(16:0) |
Phospholipid metabolism | 1.41 | ↓* |
| 50.36–282.2789 | [M + H]+ | C18H36NO | 282.2797 | −2.8 | Oleamide | Fatty acid metabolism | 1.36 | ↑* |
| 54.32–306.2781 | [M + Na]+ | C18H37NONa | 306.2773 | 2.6 | Stearamide | Fatty acid metabolism | 1.25 | ↑* |
| 41.48–542.3242 | [M + Na]+ | C26H50NO7PNa | 542.3223 | 3.5 | LysoPC(18:2) |
Phospholipid metabolism | 1.22 | ↑* |
| 3.46–204.0633 | [M + Na]+ | C9H11NO3Na | 204.0637 | −2.0 | L-Tyrosine | Amino acids metabolism | 1.25 | ↑* |
| 12.74–188.0720 | [M + H]+ | C11H10NO2 | 188.0712 | 4.3 | Indoleacrylic acid | Amino acids metabolism | 1.18 | ↑* |
| 33.21–299.2577 | [M + H]+ | C18H35O3 | 299.2586 | −3.0 | 3-Keto stearic acid | Fatty acid metabolism | 1.12 | ↑* |
| 41.16–590.3211 | [M + Na]+ | C30H50NO7PNa | 590.3223 | −2.0 | LysoPC(22:6) |
Phospholipid metabolism | 1.09 | ↑* |
| 52.82–414.2992 | [M − 2H2O + H]+ | C26H40NO3 | 414.3008 | −3.9 | GCDCA | Cholesterol metabolism | 1.06 | ↓* |
| 38.20–468.3079 | [M + H]+ | C22H47NO7P | 468.3090 | −2.3 | LysoPC(14:0) |
Phospholipid metabolism | 1.04 | ↓* |
| 3.02–118.0861 | [M + H]+ | C5H12NO2 | 118.0868 | −5.9 | L-Valine | Amino acids metabolism | 1.02 | ↑* |
| 41.17–566.3817 | [M + H]+ | C28H57NO8P | 566.3822 | −0.9 | PC(20:0) |
Phospholipid metabolism | 1.01 | ↓* |
|
||||||||
| Negative mode | ||||||||
| 55.71–363.2553 | [M + CH3COO]− | C22H35O4 | 363.2535 | 5.0 | Arachidonic acid | Fatty acid metabolism | 1.97 | ↑* |
| 3.21–114.0551 | [M − H]− | C5H8NO2 | 114.0555 | −3.5 | L-Proline | Amino acids metabolism | 1.94 | ↑* |
| 38.97–378.2416 | [M − H]− | C18H37NO5P | 378.2409 | 1.9 | Sphingosine-1-phosphate | Sphingolipid metabolism | 1.87 | ↓* |
| 20.10–832.5835 | [M − H]− | C48H83NO8P | 832.5856 | −2.5 | PC(40:6) |
Phospholipid metabolism | 1.42 | ↑* |
| 50.74–280.2649 | [M − H]− | C18H34NO | 280.2640 | 3.2 | Oleamide | Fatty acid metabolism | 1.41 | ↑* |
| 32.84–335.2238 | [M − H]− | C20H31O4 | 335.2222 | 4.8 | Leukotriene B4 | Fatty acid metabolism | 1.35 | ↓* |
| 50.37–560.3336 | [M − H]− | C28H51NO8P | 560.3352 | −2.9 | PC(20:2) |
Phospholipid metabolism | 1.31 | ↑* |
| 50.41–508.3417 | [M − H]− | C25H51NO7P | 508.3403 | 2.8 | LysoPC(17:0) |
Phospholipid metabolism | 1.28 | ↓* |
| 44.95–526.2759 | [M + CH3COO]− | C23H45NO10P | 526.2781 | −4.2 | PC(13:0) |
Phospholipid metabolism | 1.27 | ↓* |
| 27.81–381.2628 | [M − H]− | C22H37O5 | 381.2641 | −3.4 | 20-Ethyl PGF2α | Fatty acid metabolism | 1.23 | ↓* |
| 12.58–203.0831 | [M − H]− | C11H11N2O2 | 203.0821 | 4.9 | Tryptophan | Amino acids metabolism | 1.19 | ↑* |
| 23.45–754.5403 | [M − H]− | C42H77NO8P | 754.5387 | 2.1 | PC(34:3) |
Phospholipid metabolism | 1.13 | ↑* |
| 55.77–282.2793 | [M − H]− | C18H36NO | 282.2797 | −1.4 | Stearamide | Fatty acid metabolism | 1.10 | ↑* |
| 48.22–254.2475 | [M − H]− | C16H32NO | 254.2484 | −3.5 | Palmitic amide | Fatty acid metabolism | 1.07 | ↑* |
| 34.54–391.2851 | [M − C2H4NO]− | C24H39O4 | 391.2848 | 0.8 | GDCA | Cholesterol metabolism | 1.06 | ↓* |
| 43.55–480.3094 | [M − H]− | C23H47NO7P | 480.3090 | 0.3 | LysoPC(15:0) |
Phospholipid metabolism | 1.04 | ↓* |
| 44.95–556.3043 | [M − H]− | C28H47NO8P | 556.3039 | 0.7 | PC(20:4) |
Phospholipid metabolism | 1.03 | ↑* |
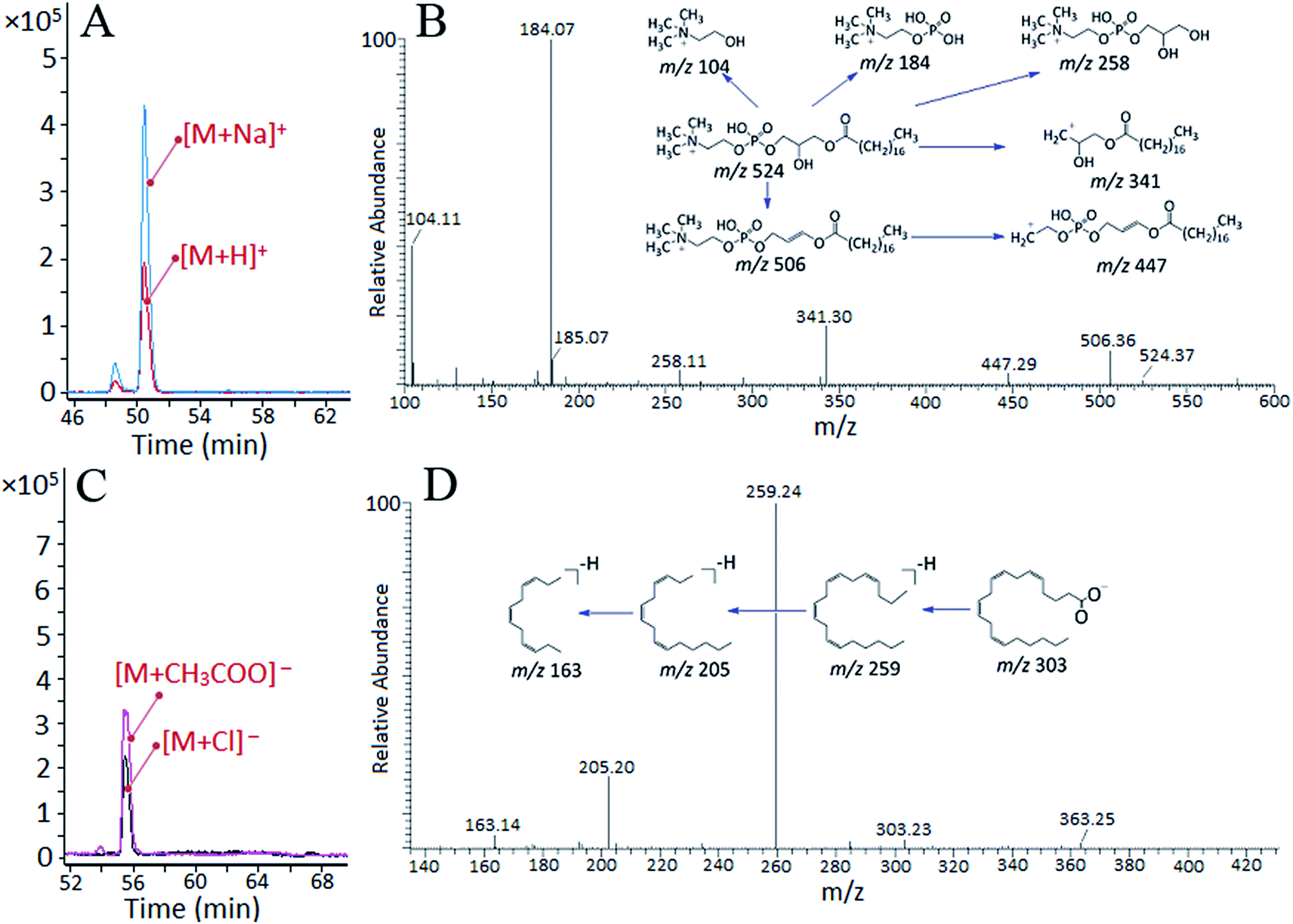 | ||
| Fig. 4 (A) EICs for lysoPC(18:0) ionized in positive ion mode with two different adducts: [M + H]+ and [M + Na]+. (B) MS/MS spectra of lysoPC(18:0) and its fragmentation behavior. (C) EICs for arachidonic acid ionized in negative ion mode with two different adducts: [M + CH3COO]− and [M + Cl]−. (D) MS/MS spectra of arachidonic acid and its fragmentation behavior. | ||
The metabolites profiling altered by AHS was found to be similar between serum and plasma. However, we observed that the increased phe–phe levels in AHS rat serum were therefore unlikely to reflect AHS-associated biological processes in vivo. In the plasma untargeted metabolomics study, the metabolites altered by AHS mainly involved in phospholipid metabolism (increased levels of PC(13:0), PC(20:0), PC(20:2), PC(20:4), PC(40:6); decreased levels of lysoPC(14:0), lysoPC(15:0), lysoPC(16:0), lysoPC(17:0), lysoPC(18:0), lysoPC(18:1) and increased levels of lysoPC(18:2), lysoPC(20:3), lysoPC(22:6)), amino acids metabolism (increased levels of L-proline, tryptophan, L-valine, L-tyrosine, L-leucine, indoleacrylic acid, phenylpyruvic acid and phenylalanine), fatty acid metabolism (increased levels of 3-keto stearic acid, oleamide, stearamide, palmitic amide, arachidonic acid and decreased levels of prostaglandin A1 (PGA1) methyl ester, 20-ethyl PGE2, 20-ethyl PGF2α, leukotriene B4), cholesterol metabolism (decreased levels of GDCA, GCDCA) and sphingolipid metabolism (decreased levels of sphingosine-1-phosphate).
3.4 Analysis of potential metabolite biomarkers of AHS
4. Conclusions
In the present work, a reliable untargeted metabolomic method based on LC-TOF/MS and LC-MS/MS was proposed and applied to rapidly analyze metabolic alterations linked to incident AHS and interrogated serum and plasma in order to identify overall differences in the metabolites levels, investigated which of the two matrices was more suitable for the discovery of endogenous biomarkers. As a result, metabolite level differences between serum and plasma samples in normal rats were mainly related to 8 LysoPCs, 3 PCs, 2 heterocyclic compounds, 7 PUFAs and their oxFAs. Notably, 7 characteristic peptides were observed only in serum. All the altered metabolites were associated with clotting cascade reaction, indicating that serum conceals the changes in metabolome that have occurred in vitro, thus obscuring variations between specimens. The results demonstrated that, compared with serum, heparin plasma which represented the original properties of the blood sample was a more suitable matrix to explore potential chemical markers in vivo. In the plasma untargeted metabolomics study, the metabolic alterations linked to AHS were mainly involved in phospholipid metabolism, amino acids metabolism, fatty acid metabolism, cholesterol metabolism and sphingolipid metabolism. These findings provide a systematic view of metabolic alterations linked to AHS, demonstrating the untargeted metabonomic method was a robust method for examining the molecular mechanisms of disease.Acknowledgements
We would like to express our gratitude to National Natural Science Foundation of China (No. 81274063), Technology Innovation Program for post-graduate of Jiangsu province (No. KYLX_0619) and a Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions. We also wish to thank Kaiwen Luo, Juanjuan Wang for assistance with the experiments.References
- G. J. Patti, O. Yanes and G. Siuzdak, Nat. Rev. Mol. Cell Biol., 2012, 13, 263–269 CrossRef CAS PubMed.
- N. Zamboni, A. Saghatelian and G. J. Patti, Mol. Cell, 2015, 58, 699–706 CrossRef CAS PubMed.
- K. Contrepois, L. H. Jiang and M. Snyder, Mol. Cell. Proteomics, 2015, 14, 1684–1695 CAS.
- W. B. Dunn, D. Broadhurst, P. Begley, E. Zelena, S. Francis-McIntyre, N. Anderson, M. Brown, J. D. Knowles, A. Halsall, J. N. Haselden, A. W. Nicholls, I. D. Wilson, D. B. Kell, R. Goodacre and The Human Serum Metabolome (HUSERMET) Consortium, Nat. Protoc., 2011, 6, 1060–1083 CrossRef CAS PubMed.
- O. Beckonert, H. C. Keun, T. M. D. Ebbels, J. G. Bundy, E. Holmes, J. C. Lindon and J. K. Nicholson, Nat. Protoc., 2007, 2, 2692–2703 CrossRef CAS PubMed.
- L. An, Q. S. Shi and F. Feng, RSC Adv., 2015, 5, 84048–84055 RSC.
- E. Holmes, A. Wijeyesekera, S. D. Taylor-Robinson and J. K. Nicholson, Nat. Rev. Gastroenterol. Hepatol., 2015, 12, 458–471 CrossRef PubMed.
- Z. J. Zhu, A. W. Schultz, J. H. Wang, C. H. Johnson, S. M. Yannone, G. J. Patti and G. Siuzdak, Nat. Protoc., 2013, 8, 451–460 CrossRef CAS PubMed.
- M. Raro, M. Ibáñez, R. Gil, A. Fabregat, E. Tudela, K. Deventer, R. Ventura, J. Segura, J. Marcos, A. Kotronoulas, J. Joglar, M. Farré, S. Yang, Y. Xing, P. Van Eenoo, E. Pitarch, F. Hernández, J. V. Sancho and Ó. J. Pozo, Anal. Chem., 2015, 87, 8373–8380 CrossRef CAS PubMed.
- X. Y. Yu, J. Luo, L. J. Chen, C. X. Zhang, R. T. Zhang, Q. Hu, S. L. Qiao and L. Li, RSC Adv., 2015, 5, 69800–69812 RSC.
- G. Szabo, Gastroenterology, 2015, 148, 30–36 CrossRef CAS PubMed.
- H. Fernando, K. K. Bhopale, S. Kondraganti, B. S. Kaphalia and G. A. S. Ansari, Toxicol. Appl. Pharmacol., 2011, 255, 127–137 CrossRef CAS PubMed.
- S. Manna, M. Thompson and F. Gonzalez, in Biological Basis of Alcohol-Induced Cancer, ed. V. Vasiliou, S. Zakhari, H. K. Seitz and J. B. Hoek, Springer International Publishing, 2015, vol. 815, ch. 13, pp. 217–238 Search PubMed.
- M. Jaremek, Z. Yu, M. Mangino, K. Mittelstrass, C. Prehn, P. Singmann, T. Xu, N. Dahmen, K. M. Weinberger, K. Suhre, A. Peters, A. Doring, H. Hauner, J. Adamski, T. Illig, T. D. Spector and R. Wang-Sattler, Transl. Psychiatry, 2013, 3, e276 CrossRef CAS PubMed.
- H. J. Issaq, Z. Xiao and T. D. Veenstra, Chem. Rev., 2007, 107, 3601–3620 CrossRef CAS PubMed.
- M. Ishikawa, K. Maekawa, K. Saito, Y. Senoo, M. Urata, M. Murayama, Y. Tajima, Y. Kumagai and Y. Saito, PLoS One, 2014, 9, e91806 Search PubMed.
- D. C. Wedge, J. W. Allwood, W. Dunn, A. A. Vaughan, K. Simpson, M. Brown, L. Priest, F. H. Blackhall, A. D. Whetton, C. Dive and R. Goodacre, Anal. Chem., 2011, 83, 6689–6697 CrossRef CAS PubMed.
- W. Zhong, W. L. Zhang, Q. Li, G. X. Xie, Q. Sun, X. H. Sun, X. B. Tan, X. G. Sun, W. Jia and Z. X. Zhou, J. Hepatol., 2015, 62, 1375–1381 CrossRef CAS PubMed.
- T. Barri and L. O. Dragsted, Anal. Chim. Acta, 2013, 768, 118–128 CrossRef CAS PubMed.
- R. Tautenhahn, K. Cho, W. Uritboonthai, Z. J. Zhu, G.J. Patti and G. Siuzdak, Nat. Biotechnol., 2012, 30, 826–828 CrossRef CAS PubMed.
- H. Gowda, J. Ivanisevic, C. H. Johnson, M. E. Kurczy, H. P. Benton, D. Rinehart, T. Nguyen, J. Ray, J. Kuehl, B. Arevalo, P. D. Westenskow, J. H. Wang, A. P. Arkin, A. M. Deutschbauer, G. J. Patti and G. Siuzdak, Anal. Chem., 2014, 86, 6931–6939 CrossRef CAS PubMed.
- M. Garcia-Aloy, R. Llorach, M. Urpi-Sarda, O. Jáuregui, D. Corella, M. Ruiz-Canela, J. Salas-Salvadó, M. Fitó, E. Ros, R. Estruch and C. Andres-Lacueva, Mol. Nutr. Food Res., 2015, 59, 212–220 CAS.
- P. A. Vorkas, G. Isaac, M. A. Anwar, A. H. Davies, E. J. Want, J. K. Nicholson and E. Holmes, Anal. Chem., 2015, 87, 4184–4193 CrossRef CAS PubMed.
- A. M. Wheelock and C. E. Wheelock, Mol. BioSyst., 2013, 9, 2589–2596 RSC.
- H. Wu, X. Li, X. Yan, L. An, K. Luo, M. Shao, Y. Jiang, R. Xie and F. Feng, J. Pharm. Biomed. Anal., 2015, 115, 315–322 CrossRef CAS PubMed.
- D. S. Wishart, T. Jewison, A. C. Guo, M. Wilson, C. Knox, Y. F. Liu, Y. Djoumbou, R. Mandal, F. Aziat, E. Dong, S. Bouatra, I. Sinelnikov, D. Arndt, J. G. Xia, P. Liu, F. Yallou, T. Bjorndahl, R. Perez-Pineiro, R. Eisner, F. Allen, V. Neveu, R. Greiner and A. Scalbert, Nucleic Acids Res., 2013, 41, D801–D807 CrossRef CAS PubMed.
- E. Fahy, M. Sud, D. Cotter and S. Subramaniam, Nucleic Acids Res., 2007, 35, W606–W612 CrossRef PubMed.
- R. Dallmann, A. U. Viola, L. Tarokh, C. Cajochen and S. A. Brown, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 2625–2629 CrossRef CAS PubMed.
- S. Bijlsma, L. Bobeldijk, E. R. Verheij, R. Ramaker, S. Kochhar, I. A. Macdonald, B. van Ommen and A. K. Smilde, Anal. Chem., 2006, 78, 567–574 CrossRef CAS PubMed.
- R. R. Miles, R. F. Roberts, A. R. Putnam and W. L. Roberts, Clin. Chem., 2004, 50, 1704–1706 CAS.
- H. Tammen, L. Schulte, R. Hess, C. Menzel, M. Kellmann, T. Mohring and P. Schulz-Knappe, Proteomics, 2005, 5, 3414–3422 CrossRef CAS PubMed.
- O. Teahan, S. Gamble, E. Holmes, J. Waxman, J. K. Nicholson, C. Bevan and H. C. Keun, Anal. Chem., 2006, 78, 4307–4318 CrossRef CAS PubMed.
- L. S. Liu, J. Y. Aa, G. J. Wang, B. Yan, Y. Zhang, X. W. Wang, C. Y. Zhao, B. Cao, J. A. Shi, M. J. Li, T. A. Zheng, Y. T. Zheng, G. Hao, F. Zhou, J. G. Sun and Z. M. Wu, Anal. Biochem., 2010, 406, 105–112 CrossRef CAS PubMed.
- J. Aoki, A. Taira, Y. Takanezawa, Y. Kishi, K. Hama, T. Kishimoto, K. Mizuno, K. Saku, R. Taguchi and H. Arai, J. Biol. Chem., 2002, 277, 48737–48744 CrossRef CAS PubMed.
- W. E. Wung and S. B. Howell, Clin. Chem., 1980, 26, 1704–1708 CAS.
- C. S. Lieber, S. J. Robins, J. J. Li, L. M. Decarli, K. M. Mak, J. M. Fasulo and M. A. Leo, Gastroenterology, 1994, 106, 152–159 CAS.
- A. A. Chirkin, N. Y. Konevalova, I. N. Grebennikov, V. A. Kulikov, Y. V. Saraev, V. U. Buko, I. A. Chirkina, E. O. Danchenko and K. J. Gundermann, Addict. Biol., 1998, 3, 65–70 CrossRef CAS PubMed.
- J. Chang, J. H. Musser and H. McGregor, Biochem. Pharmacol., 1987, 36, 2429–2436 CrossRef CAS PubMed.
- S. Pyne, K. C. Kong and P. I. Darroch, Semin. Cell Dev. Biol., 2004, 15, 491–501 CrossRef CAS PubMed.
- A. I. Cederbaum, Y. K. Lu and D. F. Wu, Arch. Toxicol., 2009, 83, 519–548 CrossRef CAS PubMed.
- C. S. Lieber, J. Hepatol., 2000, 32, 113–128 CrossRef CAS PubMed.
- X. Tang, E. M. Edwards, B. B. Holmes, J. R. Falck and W. B. Campbell, Am. J. Physiol.: Heart Circ. Physiol., 2005, 290, H37–H45 CrossRef PubMed.
- H. K. Min, A. Kapoor, M. Fuchs, F. Mirshahi, H. P. Zhou, J. Maher, J. Kellum, R. Warnick, M. J. Contos and A. J. Sanyal, Cell Metab., 2012, 15, 665–674 CrossRef CAS PubMed.
- Y. Liu, S. Y. Saiyan, T. Y. Men, H. Y. Gao, C. Wen, Y. Liu, X. Zhou, C. T. Wu, L. S. Wang and C. P. Cui, J. Pathol., 2013, 230, 365–376 CrossRef CAS PubMed.
- H. Ikeda, N. Watanabe, I. Ishii, T. Shimosawa, Y. Kume, T. Tomiya, Y. Inoue, T. Nishikawa, N. Ohtomo, Y. Tanoue, S. Iitsuka, R. Fujita, M. Omata, J. Chun and Y. Yatomi, J. Lipid Res., 2008, 50, 556–564 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra27910k |
| This journal is © The Royal Society of Chemistry 2016 |