
Amide-containing luminescent metal–organic complexes as bifunctional materials for selective sensing of amino acids and reaction prompting†
Pengyan Wua,
Min Jianga,
Xuefu Hub,
Jingru Wanga,
Guangjie He*c,
Ying Shia,
Yang Lia,
Wei Liua and
Jian Wang*a
aSchool of Chemistry and Chemical Engineering, Jiangsu Key Laboratory of Green Synthetic Chemistry for Functional Materials, Jiangsu Normal University, Xuzhou, Jiangsu 221116, P. R. China. E-mail: wjian@jsnu.edu.cn
bState Key Laboratory of Physical Chemistry of Solid Surfaces, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen, 361005, P. R. China
cDepartment of Forensic Medicine, Xinxiang Medical University, XinXiang, 453003, China. E-mail: guangjiehe@163.com
First published on 11th March 2016
Abstract
Two new cerium-based luminescent metal–organic complexes (MOCs), Ce–BBAS and Ce–TBAS, were achieved via self-assembly of two and three additional free amide-functionalized ligands and cerium(III) nitrate, respectively. These two MOCs exhibit different opening size and inner cavities. The smaller metal–organic triple-stranded helicate Ce–BBAS exhibits high selectivity toward aspartic acid (Asp) over other natural amino acids, with the detection limit of ca. 0.26 μM, providing the first artificial receptor that can specifically discriminate Asp over other amino acids in a luminescence enhancement manner. As a comparison, the bigger metal–organic tetrahedron Ce–TBAS doesn't reflect this nature, demonstrating size-selective sensing. In addition, these two MOCs could also work as luminescent chemosensors towards salicylaldehyde derivatives, and feature confined cavities and amide groups as a base catalytic driving force to enable the Knoevenagel condensation reactions.
Introduction
The development of molecular recognition for biologically important molecules has received considerable attention in recent years;1,2 in particular, amino acids (AAs) are important bioactive substances and they are widely used in the food, chemical and pharmaceutical industries.3,4 Among them, aspartic acid (Asp) is a major excitatory neurotransmitter in the central nervous system.5,6 An excess of Asp causes amyotrophic lateral sclerosis (ALS or Lou Gehrig's disease), stroke, and epilepsy.7 A deficiency might be associated with its metabolic disorders to manifest lung cancer at 2 μM and a typical disease like head and neck cancer at 7.0 μM (normal level: 21.0 μM).8 A practical sensor of Asp covering a wide range of detection is called for. Inspired by the structures of the natural enzymatic pocket, recent development on the selectively AAs recognition mainly depend on employing the hydrogen bonding triggers within the well-defined luminophores for their sensitive responding, since fluorescence method has got a rapid development for the high sensitivity, selectivity, fast response, spatial resolution, real time monitoring and noninvasiveness.9,10 However, all of the AAs exhibit both hydrogen bonding donors and acceptors due to the intrinsic properties of their amide groups,11 the design of highly efficient probes for selectively sensing and differentiating AAs still remains a challenge.The amide group is a fascinating functional group because it possesses two types of hydrogen-bonding sites: the –NH moiety acts as an electron acceptor and the –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group acts as an electron donor.12 Thus, the incorporation of amide groups as guest-accessible sites into the fluorophore moiety to achieve functional materials will be a good choice for such sensing application. In addition, combined with the structure features of natural amino acids, the material with the structural distinction is needed. Metal–organic complexes (MOCs), such as macrocycles and polyhedral, with discrete molecular architectures constructed through the coordination of metal ions and organic linkers, have attracted considerable attention due to their high symmetry, stability, rich chemical/physical properties and restricted cavities.13,14 Their tunable pore size and characteristic functionality that are similar to active sites in proteins suggest they may act as bright promising host matrices for molecular recognition,15 because the inherent confinement effect within their pores serves as preconcentrator to enhance the host–guest interactions, such as hydrogen bonding, π-stacking, electrostatic, and hydrophilic/lipophilic interactions, in a controllable way.16 Accordingly, design and synthesis of the metal–organic complexes incorporated with amide groups and functionalized with fluorescence properties, will be a good candidate material for natural amino acids sensing.
O group acts as an electron donor.12 Thus, the incorporation of amide groups as guest-accessible sites into the fluorophore moiety to achieve functional materials will be a good choice for such sensing application. In addition, combined with the structure features of natural amino acids, the material with the structural distinction is needed. Metal–organic complexes (MOCs), such as macrocycles and polyhedral, with discrete molecular architectures constructed through the coordination of metal ions and organic linkers, have attracted considerable attention due to their high symmetry, stability, rich chemical/physical properties and restricted cavities.13,14 Their tunable pore size and characteristic functionality that are similar to active sites in proteins suggest they may act as bright promising host matrices for molecular recognition,15 because the inherent confinement effect within their pores serves as preconcentrator to enhance the host–guest interactions, such as hydrogen bonding, π-stacking, electrostatic, and hydrophilic/lipophilic interactions, in a controllable way.16 Accordingly, design and synthesis of the metal–organic complexes incorporated with amide groups and functionalized with fluorescence properties, will be a good candidate material for natural amino acids sensing.
On the other hand, the application of metal–organic complexes as “molecular flasks” has also precipitated a surge of interest in the reactivity and property of molecules within well-defined spaces.17,18 However, most metal–organic complexes don't constitute specific interactions capable of encapsulating guest molecules and catalyzing the reactions, due to the intrinsic difficulties on the producing guest accessible sites inside the well-defined spaces.19 Herein, amide groups containing metal–organic complexes are a powerful approach to achieve functional flasks to prompt several important reactions, since amide group was often used as a base catalytic driving force.20 Through carefully incorporating amide groups functionalization of organic fluorescent moieties as guest-accessible sites within the well defined metal–organic complexes, two new cerium-based metal–organic complexes, Ce–BBAS and Ce–TBAS, with different size of the windows and cavities were achieved by self-assembly by introducing 2-folds and 3-folds amide-containing tridentate chelating sites into the fragments of the ligands, respectively (Scheme 1). In light of the amide groups on their internal surface and the cavities sieving, the sensing of natural amino acids and catalytic abilities of Knoevenagel condensations reaction were studied.
 | ||
| Scheme 1 Molecular structures of H4BBAS and H6TBAS, and constitutive/constructional fragments of the cerium based metal–organic triple-stranded helicate Ce–BBAS and metal–organic tetrahedron Ce–TBAS afforded by theoretical calculation. | ||
Results and discussion
Ligand H4BBAS was obtained by a simple Schiff-base reaction of salicylaldehyde with dimethyl 4′,4′′′-((4-bromophenyl)azanediyl)bis(([1,1′-biphenyl]-4′-carbohydrazide)) in a methanol solution. Reaction of ligands H4BBAS with Ce(NO3)3·6H2O in the presence of KOH in DMF solution, and diffusing with methanol affords new compounds Ce–BBAS. ESI-MS spectrum of Ce–BBAS in DMF/CH3OH solution exhibited an intense peaks at m/z = 890.11 with the isotopic distribution patterns separated by 0.33 dalton (Fig. 1), that signal was assignable to the species of [Ce(IV)2(BBAS)2(HBBAS)]3−, implying the formation of [M2L3] type metal–organic species and its substantial stability in solution. The Ce 3d core level XPS spectrum exhibited a distinct band at about 916 eV, which was assignable beside the peaks at around 880–890 and 895–910 distinct regions. The unique peak was assigned to 4f0 orbital transitions,21 suggesting the presence of the CeIV state in compound Ce–BBAS. The bonding of the ligands to the metal ions was also identified by the relatively broadened and shifted resonance signals in 1H NMR spectra (Fig. 1). Precisely, the disappearance of the phenolic proton signal at ∼11.30 ppm and the significant upfield shift of aromatic protons in the phenol ring suggested the coordination occurred between the deprotonated phenolic groups and the metal ions. The downfield shift from 12.21 to 13.15 ppm and the reduction of the proton portion of the amide signal were indicative of the coordination of amide groups to the metal ions and the partial deprotonation of amide groups during the coordination process. Thus, Ce–BBAS is likely to be similar to that of the oxydibenzene derived bis-tridentate analogue Ce–DBOS reported in the literature,22 implying the formation of metal–organic triple-stranded helicate. Meanwhile, we also carried out computational studies by using DFT at the UB3LYP/6-31G level of theory23 to gain the sight into the optimized geometries of Ce–BBAS, which supported the formation of the metal–organic triple-stranded helicate structure. | ||
| Fig. 1 ESI-MS of Ce–BBAS (0.1 mM) in DMF/CH3OH (top) and the relative 1H NMR spectra of H4BBAS and Ce–BBAS in DMSO-d6 (bottom). | ||
The absorption of ligand H4BBAS in DMF/H2O = 8/2 exhibited an intense band about 335 nm, assignable to the transitions associated with the phenol groups.24 Whereas compound Ce–BBAS exhibited an intensive absorption at about 350 nm, the significant shifts and intensity variations compared to those of free ligand, indicated the deprotonation of phenol groups and the metal–ligand interactions (Fig. S5†). When excited at 350 nm, compound Ce–BBAS (15 μM) exhibited an emission band centred at 460 nm in DMF/water 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 (v/v). Upon the addition of Asp, the luminescence intensity of Ce–BBAS enhanced gradually with the increasing concentration of the guest, and exhibited a 4.3-fold increase when the amount of the added Asp reached to 0.13 mM. Hill-plot25 of the titration curves well confirmed a 1:1 stoichiometric host–guest behavior, with an associate constant (logKass) of 4.03 ± 0.11 (Fig. 2a and S11†). The fluorescence intensity of the solution was nearly proportional to the amount of Asp in the range of 0 to 4 μM, suggesting that Ce–BBAS was potentially useful for quantitative determination of Asp (Fig. 2a, insert). The detection limit was established at or below 0.26 μM according to the 3δ IUPAC criteria, which is much sensitive than the deficiency of Asp content in human cells for the cancer occurred. The addition of Asp did not cause any obvious change in the UV-vis spectrum of Ce–BBAS (Fig. S5†). Based on the observed insensitivity of the UV-vis absorptions of Ce–BBAS upon addition of Asp, we may attribute the observed emission enhancement to the rigidification of the structure induced by the interaction with Asp. This structural rigidification would render vibrational deactivation pathways less efficient thereby leading to an increase in emission quantum yield.26 Therefore, we believe that Ce–BBAS is interesting in the development of new measurement in clinical applications, especially in manifest lung cancer, head and neck cancer detection.
:2 (v/v). Upon the addition of Asp, the luminescence intensity of Ce–BBAS enhanced gradually with the increasing concentration of the guest, and exhibited a 4.3-fold increase when the amount of the added Asp reached to 0.13 mM. Hill-plot25 of the titration curves well confirmed a 1:1 stoichiometric host–guest behavior, with an associate constant (logKass) of 4.03 ± 0.11 (Fig. 2a and S11†). The fluorescence intensity of the solution was nearly proportional to the amount of Asp in the range of 0 to 4 μM, suggesting that Ce–BBAS was potentially useful for quantitative determination of Asp (Fig. 2a, insert). The detection limit was established at or below 0.26 μM according to the 3δ IUPAC criteria, which is much sensitive than the deficiency of Asp content in human cells for the cancer occurred. The addition of Asp did not cause any obvious change in the UV-vis spectrum of Ce–BBAS (Fig. S5†). Based on the observed insensitivity of the UV-vis absorptions of Ce–BBAS upon addition of Asp, we may attribute the observed emission enhancement to the rigidification of the structure induced by the interaction with Asp. This structural rigidification would render vibrational deactivation pathways less efficient thereby leading to an increase in emission quantum yield.26 Therefore, we believe that Ce–BBAS is interesting in the development of new measurement in clinical applications, especially in manifest lung cancer, head and neck cancer detection.
 | ||
| Fig. 2 (a) Family of the luminescence spectra of Ce–BBAS (15 μM) upon addition of Asp up to 0.13 mM. The inset picture shows the fluorescence at 460 nm of compound Ce–BBAS as a function of Asp concentration, each error bar represents the data range. (b) Luminescent responses of Ce–BBAS (15 μM) to various amino acids in DMF/H2O (λex = 350 nm); the red bars represent the emission intensities of Ce–BBAS in the presence of 0.26 mM of the amino acids. The blue bars represent the change of the emission that occurs upon the subsequent addition of 0.13 mM of Asp to the above solution. | ||
To determine whether Ce–BBAS acts as a highly selective chemosensor for Asp, the luminescence responses towards other 19 kinds of natural amino acids, including Pro, Leu, Val, Met, Lys, Tyr, Ile, Thr, Ser, Trp, His, Arg, Gln, Ala, Gly, Asn, Glu, Cys and Phe (Fig. 2 top) were added respectively into a solution of Ce–BBAS under the same conditions. The addition of those species did not cause any significant luminescence changes. The competition experiments revealed that the Asp-induced luminescence response is unaffected in the background of 2 equiv. of other 19 kinds of amino acids, indicating that Ce–BBAS has a remarkable selectivity for Asp (Fig. 2b). And Ce–BBAS was described as the first artificial receptor that can specifically discriminate Asp over other amino acids in a luminescent enhancement manner.
The clarification of the host–guest interaction between Asp and Ce–BBAS was discussed. In the presence of one molar equivalent of Asp, ESI-MS spectra of Ce–BBAS in a N,N-dimethylformamide solution exhibited a new peak at 934.68 relative to its original one, assignable to the host–guest complexation species [(Ce–BBAS)⊃Asp]3− (Fig. 3 top). These results supported the 1:1 stoichiometry of the host–guest behavior. Upon addition of other natural amino acids instead of Asp, no host–guest complex species were detected in the ESI-MS spectra under the same experimental conditions, which also suggested a selective recognition of Asp by compound Ce–BBAS in solution. 1H NMR spectroscopy of Ce–BBAS (10 mM) in a DMSO-d6 (dimethyl sulfoxide) solution (Fig. 3 bottom), upon addition of one molar equivalent of Asp, exhibited significant downfield shifts of protons of the Asp with the Δδ sequence as: Δδ(H3) = 0.77 > Δδ(H2) = 0.46 > Δδ(H1) = 0.36 ppm, which demonstrated that Asp molecule was encapsulated within the cage, meanwhile, the appearance of a new peak at about 16.18 ppm might be an indicator of the formation of hydrogen bonds by the amide groups acting as donors, leading to the fluorescent changes.27 Furtherly, the molecular geometry calculations of the host–guest complex [(Ce–BBAS)⊃Asp] was carried out, and as shown in Fig. 3 top insert, thus suggesting formation of hydrogen–bonding interaction of the amide group with Asp; this result is also in good agreement with our experimental results.
 | ||
| Fig. 3 (Top) ESI-MS of Ce–BBAS (0.1 mM) contain one eq. Asp in DMF–CH3OH solution. (Bottom) Partial 1H NMR spectra of (a) Ce–BBAS (10 mM) in DMSO-d6, (b) free Ce–BBAS upon the addition of Asp (10 mM, the insert showing the chemical shift at about 16.18 ppm), and (c) Asp itself. | ||
In order to explore whether the other factors affect the luminescence response of Ce–BBAS toward Asp, a Ce-based bigger compound Ce–TBAS with tri-tridentate chelating units was constructed. Diffusing methanol into a DMF solution containing H6TBAS (Scheme 2) with Ce(NO3)3·6H2O in the presence of KOH affords new compounds Ce–TBAS. The structure was confirmed by ESI-MS, 1H NMR and theoretical calculation (Fig. S3 and S4† and Scheme 1 bottom), implying the formation of [Ce4L4] type tetrahedron complexes and its substantial stability in solution. Similar to Ce–BBAS, Ce–TBAS (15 μM) also exhibited bright blue emission at about 460 nm when excited at 350 nm. Upon the addition of Asp up to 0.13 mM into Ce–TBAS DMF/H2O = 8/2 mixture solution, the fluorescence intensity increased up to 1.2-fold. Nevertheless, the same amounts of other amino acids species caused the almost same fluorescence change (1.13-fold ∼ 1.38-fold) compared to the addition of Asp. Like the results by He et al.,28 the cavities and window size of Ce–TBAS are too large to confine and sieve some specific amino acids. So that the high selectivity between Ce–BBAS and Asp is not only attributed to the weak interaction of hydrogen bonds, but also the size restriction of the triple helical in the recognition process.
 | ||
| Scheme 2 The synthesis route of H6TBAS. | ||
Free amide groups of Ce–BBAS and Ce–TBAS could act as basic catalysis sites to promote the Knoevenagel condensation reaction that requires the formation of an active methylene anion under weak base-catalyzed conditions.29 To confirm the selective accommodation and activation of reactants by Ce–BBAS and Ce–TBAS, selective fluorescent response experiments were firstly performed to identify the substrates of Ce–BBAS and Ce–TBAS. The substrates chosen for the reaction were salicylaldehyde (SA), 1-hydroxy-2-naphthaldehyde (1-OH-2-NPA) or 4-(benzyloxy)-2-hydroxy-benzaldehyde (4-BO-2-OHBA) (Fig. 4). For Ce–TBAS, their fluorescence intensity decreased gradually with the increasing concentration of these three substrates, and a Hill-plot profile of the titration curves demonstrated a 1:1 stoichiometric host–guest behavior with the association constant (logKass) calculated as 3.25, 3.08 and 2.73 for SA, 1-OH-2-NPA and 4-BO-2-OHBA, respectively. Whereas, in case of the smaller complex Ce–BBAS, only SA addition gave similar association constant (logKass) of 3.05 as Ce–TBAS, other aldehydes, such as 1-OH-2-NPA or 4-BO-2-OHBA in excess just caused a spot of luminescent spectroscopic variations of Ce–BBAS. These results indicated that only the molecules having suitable size could enter into the cavities and be activated by the amide groups in Ce–BBAS.
 | ||
| Fig. 4 (a) Family of emission spectra of Ce–TBAS (15 μM) upon the addition of salicylaldehyde (SA) up to 0.3 mM. (b) Luminescence responses of Ce–TBAS and Ce–BBAS (15 μM) upon addition of aldehydes (0.3 mM) interested. Intensities were recorded at 460 nm, respectively, excitation at 350 nm. | ||
Knoevenagel condensation reactions were employed with a 1:2.4 mol ratio of the selected aromatic aldehyde and malononitrile in DMSO at room temperature. As shown in Table 1, the loading of 2 mol% Ce–TBAS led to a more than 60% conversion for the selected aldehydes. In contrast, SA produces 85% conversion of the adduct catalyzed by Ce–BBAS, whereas the conversion yield for 1-OH-2-NPA and 4-BO-2-OHBA are reduced to 32% and 11%, respectively, under similar experimental conditions. Despite there are lots of factors influencing the conversion of the reactions, the size selective catalytic properties as well as the same sequence of the reactivity and the response efficiency partly demonstrated that the recognition process seems the important step for these Knoevenagel condensations reaction.30 Moreover, a number of control experiments were carried out. The use of H4BBAS or H6TBAS themselves as the catalyst exhibited the conversion of below 30% but no significant difference in the conversion for different substrates. N-Benzylbenzamide containing amide could also prompt Knoevenagel condensation reaction in homogenous manner with the conversion of 28% for SA. In addition, using Ce(NO3)3·6H2O as the catalyst, it caused no conversion under the same experimental conditions. That set of experiments demonstrates that the proper cavities and the presence of amide groups played crucial roles in the Knoevenagel condensation reactions.
|
|||||
|---|---|---|---|---|---|
| Entry | Substrates | Ce–TBAS (%) | H6TBAS | Ce–BBAS (%) | H4BBAS |
| a Reaction conditions: reactions were performed with 2 mmol substrates in 10 mL of DMSO using 0.04 mmol, 2 mol% of Ce–TBAS, Ce–BBAS or ligand (0.16 mmol H6TBAS and 0.12 mmol H4BBAS) for 3 h. The conversions were determined by 1H NMR, based on starting materials.b Reaction conditions: reactions were performed with 2 mmol substrates, 2 mmol Asp in 10 mL of DMSO using 0.04 mmol, 2 mol% of Ce–BBAS for 3 h. | |||||
| 1 |  |
88 | 25 | 85 (<3)b | 24 |
| 2 | 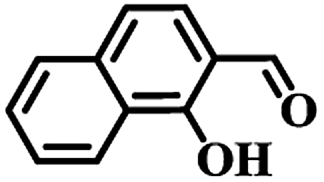 |
81 | 23 | 32 (<3) | 23 |
| 3 | 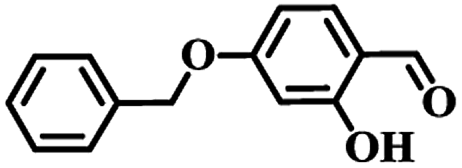 |
62 | 22 | 11 (<2) | 21 |

To further validate that the Knoevenagel condensations reaction occurred within the cavity of Ce–BBAS through a normal homogeneous system, the inhibition of the reaction was performed through the addition of non-reactive species Asp. Since Asp exhibited larger association constant for recognition by Ce–BBAS than aldehydes, Ce–BBAS was more inclined to interact with Asp. As can be expected, when adding one equiv. of Asp, the Knoevenagel condensations reaction of SA by the Ce–BBAS system was stopped. All of the experimental results strongly suggest that the catalytic reactions occurred within the inner cavities of Ce–BBAS.
Conclusions
In conclusion, two new cerium-based metal–organic complexes, Ce–BBAS and Ce–TBAS functionalized with six or twelve additional free amide groups in the cavity were successfully achieved. With the amide groups as the excellent signal responding communications and interaction sites, the size-selective amino acids sensing was discussed. The smaller metal–organic triple-stranded helicate Ce–BBAS exhibits high selectivity toward aspartic acid with the low detection limit (ca. 0.26 μM), providing the first artificial receptor for Asp sensing among twenty amino acids in a luminescent enhancement manner. Detailed experiments demonstrated that sensing of Asp by Ce–BBAS was attributed to cooperation of hydrogen bonding between Asp molecules and amide groups and size matching. In addition, the fluorescent response toward aldehydes of Ce–BBAS is also size-selective, thus driving an efficiently active process for the Knoevenagel condensations reactions in a size-selective fashion. These remarkable results may spark real application prospects of multifunctional materials.Experimental
General information
All chemicals were of reagent grade quality obtained from commercial sources and used without further purification. The elemental analyses of C, H and N were performed on a Vario EL III elemental analyzer. 1H NMR and 13C NMR spectra was measured on a Varian INOVA 400 M and 100 M spectrometer, respectively. ESI mass spectra were carried out on a HPLC-Q-Tof MS spectrometer using methanol as mobile phase. X-ray photoelectron spectroscopy (XPS) were performed with a Thermo VG ESCALAB MK2 system operating in the parallel data acquisition mode using monochromatic Al Kα radiation (hν = 1486.68 eV; spot size 400 μm). The structure optimizations were carried out by employing the unrestricted B3LYP hybrid functional using the Gaussian 09, package.31 UV-vis spectra were measured on a JASCO V-530 spectrometer. The solution fluorescent spectra were measured on F-4600 spectrometer (Hitachi). Both excitation and emission slit widths were 2.5 nm. The solution of Ce–TBAS and Ce–BBAS was prepared in DMF directly. And the high concentration stock solutions of amino acids and aldehyde (2.0 × 10−2 M) were prepared directly in water and DMF solvents, respectively, the intensity was recorded at 460 nm, excitation at 350 nm.Synthesis of H6TBAS
N–), 143.65 (CAr), 141.74 (CAr), 137.35 (CAr), 132.35 (CAr), 131.97 (CAr), 130.51 (CAr), 130.05 (CAr), 129.88 (CAr), 128.97 (CAr), 127.18 (CAr), 119.90 (CAr), 119.24 (CAr), 116.97 (CAr). Anal. calc. for C60H45N7O6: H 4.72, C 75.06, N 10.21; found: H 4.73, C 75.12, N 10.20. TOF-MS-ES(−) calcd for C60H45N7O6 958.3353, found 958.3423.Synthesis of H4BBAS
 | ||
| Scheme 3 The synthesis route of H4BBAS. | ||
N–), 142.80 (CAr), 141.78 (CAr), 137.52 (CAr), 133.77 (CAr), 132.74 (CAr), 131.97 (CAr), 131.35 (CAr), 130.05 (CAr), 130.03 (CAr), 128.96 (CAr), 127.59 (CAr), 127.19 (CAr), 120.14 (CAr), 119.91 (CAr), 119.24 (CAr), 118.93 (CAr), 117.06 (CAr). Anal. calc. for C60H45N7O6: H 4.28, C 69.00, N 8.75; found: H 4.27, C 69.12, N 8.76. TOF-MS-ES(−) calcd for C46H34BrN5O4 798.1716, found 789.1628.Synthesis of Ce–BBAS and Ce–TBAS
Acknowledgements
This work was financial support from the NSFC (21401086, 21401087, 21371148), the Natural Science Foundation of Jiangsu Province (BK20140234), PAPD of Jiangsu Higher Education Institutions, and Start-Up Fund of Jiangsu Normal University.Notes and references
- R. W. Sinkeldam, N. J. Greco and Y. Tor, Chem. Rev., 2010, 110, 2579 CrossRef CAS PubMed; X. Chen, Y. Zhou, X. Peng and J. Yoon, Chem. Soc. Rev., 2010, 39, 2120 RSC.
- J. L. Atwood, J. E. D. Davies, D. D. MacNicol, F. Vogtle and J.-M. Lehn, Comprehensive Supramolecular Chemistry, Elsevier, Amsterdam, 1996, vol. 1–11 Search PubMed.
- S. Kubik, Chem. Soc. Rev., 2009, 38, 585 RSC; L. Mutihac, J. H. Lee, J. S. Kim and J. Vicens, Chem. Soc. Rev., 2011, 40, 2777 RSC; J. Wang, H. B. Liu, Z. Tong and C. S. Ha, Coord. Chem. Rev., 2015, 303, 139 CrossRef CAS.
- Y. Zhou and J. Yoon, Chem. Soc. Rev., 2012, 41, 52 RSC; A. C. Evans, C. Meinert, C. Giri, F. Goesmann and U. J. Meierhenrich, Chem. Soc. Rev., 2012, 41, 5447 RSC; M. S. Alaejos and F. J. G. Montelongo, Chem. Rev., 2004, 104, 3239 CrossRef CAS PubMed.
- M. Rekharsky, H. Yamamura, M. Kawai and Y. Inoue, J. Am. Chem. Soc., 2001, 123, 5360 CrossRef CAS PubMed; B. B. Prasad, A. Srivastava and M. P. Tiwari, J. Chromatogr. A, 2013, 1283, 9 CrossRef PubMed.
- B. B. Prasad and I. Pandey, Electrochim. Acta, 2013, 88, 24 CrossRef CAS.
- M. H. Baslow and T. R. Resnik, J. Mol. Neurosci., 1997, 9, 109 CrossRef CAS PubMed; J. Gao, D. Lv, H. Sun and W. Yang, J. Braz. Chem. Soc., 2009, 20, 1827 CrossRef.
- N. Psychogios, D. D. Hau, J. Peng, A. C. Guo, R. Mandal, S. Bouatra, I. Sinelnikov, R. Krishnamurthy, R. Eisner, B. Guatam, N. Young, J. Xia, C. Knox, E. Dong, P. Haung, Z. Hollander, T. L. Pedersen, S. R. Smith, F. Bamforth, R. Greiner, B. McManus, J. W. Newman and T. Goodfriend, PLoS One, 2011, 6, e16957 CAS; M. C. Dols, M. D. Lopez, C. R. Plaza, E. P. Miranda, S. G. Calle, E. V. Chamorro, I. A. Diaz, A. M. Pino, J. A. Garcia and V. G. Calderon, Oncologia, 2006, 29, 283 Search PubMed.
- M. Zhang, M. Yu, M. Zhu, M. Li, Y. Gao, L. Li, F. Li, Z. Liu, J. Zhang, D. Zhang, T. Yi and C. Huang, J. Am. Chem. Soc., 2007, 129, 10322 CrossRef CAS PubMed; C. Schmuck and U. Machon, Chem.–Eur. J., 2005, 11, 1109 CrossRef PubMed.
- A. P. de Silva, H. Q. N. Gunaratne, T. Gunnlaugsson, A. J. M. Huxley, C. P. McCoy, J. T. Rademacher and T. E. Rice, Chem. Rev., 1997, 97, 1515 CrossRef CAS PubMed; B. Valeur and I. Leray, Coord. Chem. Rev., 2000, 205, 3 CrossRef; Y. M. Yang, Q. Zhao, W. Feng and F. Y. Li, Chem. Rev., 2013, 113, 192 CrossRef PubMed; X. Q. Chen, T. Pradhan, F. Wang, J. S. Kim and J. Yoon, Chem. Rev., 2012, 112, 1910 CrossRef PubMed.
- S. S. Yoon and W. C. Still, J. Am. Chem. Soc., 1993, 115, 823 CrossRef CAS; Y. Kuroda, Y. Kato, T. Higashioji, J. Hasegawa, S. Kawanami, M. Takahashi, N. Shiraishi, K. Tanabe and H. Ogoshi, J. Am. Chem. Soc., 1995, 117, 10950 CrossRef.
- F. A. Quiocho, Pure Appl. Chem., 1989, 61, 1293 CrossRef CAS; N. K. Vyas, M. N. Vyas and F. A. Quiocho, Science, 1988, 242, 1290 Search PubMed; C. M. Lee and W. D. Kumler, J. Am. Chem. Soc., 1962, 84, 571 CrossRef; H. A. Bent, Chem. Rev., 1968, 68, 587 CrossRef; P. L. Huyskens, J. Am. Chem. Soc., 1977, 99, 2578 CrossRef.
- B. H. Northrop, Y. R. Zheng, K. W. Chi and P. J. Stang, Acc. Chem. Res., 2009, 42, 1554 CrossRef CAS PubMed; M. Yoshizawa, J. K. Klosterman and M. Fujita, Angew. Chem., Int. Ed., 2009, 48, 3418 CrossRef PubMed; T. R. Cook, R. Y. Yang and P. J. Stang, Chem. Rev., 2013, 113, 734 CrossRef PubMed; S. Zarra, D. M. Wood, D. A. Roberts and J. R. Nitschke, Chem. Soc. Rev., 2015, 44, 419 RSC.
- L. Cronin, Angew. Chem., Int. Ed., 2006, 45, 3576 CrossRef CAS PubMed; C. Schmuck, Angew. Chem., Int. Ed., 2007, 46, 5830 CrossRef PubMed; A. Lutzen, Angew. Chem., Int. Ed., 2005, 44, 1000 CrossRef PubMed; M. D. Pluth, R. G. Bergman and K. N. Raymond, Acc. Chem. Res., 2009, 42, 1650 CrossRef PubMed; K. Li, L. Y. Zhang, C. Yan, S. C. Wei, M. Pan, L. Zhang and C. Y. Su, J. Am. Chem. Soc., 2014, 136, 4456 CrossRef PubMed; B. Roy, A. K. Ghosh, S. Srivastava, P. D'Silva and P. S. Mukherjee, J. Am. Chem. Soc., 2015, 137, 11916 CrossRef PubMed.
- E. Barea, J. A. R. Navarro, J. M. Salas, M. Quiro, M. Willermann and B. Lippert, Chem.–Eur. J., 2003, 9, 4414 CrossRef CAS PubMed; S. Tashiro, M. Kobayashi and M. Fujita, J. Am. Chem. Soc., 2006, 128, 9280 CrossRef PubMed; C. J. Hastings, D. Fiedler, R. G. Bergman and K. N. Raymond, J. Am. Chem. Soc., 2008, 130, 10977 CrossRef PubMed; A. Jiménez, R. A. Bilbeisi, T. K. Ronson, S. Zarra, C. Woodhead and J. R. Nitschke, Angew. Chem., Int. Ed., 2014, 53, 4556 CrossRef PubMed.
- R. Chakrabarty, P. S. Mukherjee and P. J. Stang, Chem. Rev., 2011, 111, 6810 CrossRef CAS PubMed; J. T. Davis and G. P. Spada, Chem. Soc. Rev., 2007, 36, 296 RSC; D. Ramaiah, P. P. Neelakandan, A. K. Nair and R. R. Avirah, Chem. Soc. Rev., 2010, 39, 4158 RSC.
- C. J. Brown, F. D. Toste, R. G. Bergman and K. N. Raymond, Chem. Rev., 2015, 115, 3012 CrossRef CAS PubMed; M. Fujita, M. Tominaga, A. Hori and B. Therrien, Acc. Chem. Res., 2005, 38, 369 CrossRef PubMed; M. D. Ward, Chem. Commun., 2009, 4487 RSC; D. Fiedler, D. H. Leung, R. G. Bergman and K. N. Raymond, Acc. Chem. Res., 2005, 38, 349 CrossRef PubMed; R. W. Saalfrank, H. Maid and A. Scheurer, Angew. Chem., Int. Ed., 2008, 47, 8794 CrossRef PubMed.
- I. M. Muller and D. Moller, Angew. Chem., Int. Ed., 2005, 44, 2969 CrossRef CAS PubMed; S. Hiraoka, K. Harano, M. Shiro, Y. Ozawa, N. Yasuda, K. Toriumi and M. Shionoya, Angew. Chem., Int. Ed., 2006, 45, 6488 CrossRef PubMed; I. S. Tidmarsh, T. B. Faust, H. Adams, L. P. Harding, L. Russo, W. Clegg and M. D. Ward, J. Am. Chem. Soc., 2008, 130, 15167 CrossRef PubMed; D. Moon, S. Kang, J. Park, K. Lee, R. P. John, H. Won, G. H. Seong, Y. S. Kim, G. H. Kim, H. Rhee and M. S. Lah, J. Am. Chem. Soc., 2006, 128, 3530 CrossRef PubMed.
- M. D. Pluth, D. Fiedler, J. S. Mugridge, R. G. Bergman and K. N. Raymond, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 10438 CrossRef CAS PubMed; D. M. Vriezema, M. C. Aragone's, J. A. A. W. Elemans, J. J. L. M. Cornelissen, A. E. Rowan and R. J. M. Nolte, Chem. Rev., 2005, 105, 1445 CrossRef PubMed.
- S. Hasegawa, S. Horike, R. Matsuda, S. Furukawa, K. Mochizuki, Y. Kinoshita and S. Kitagawa, J. Am. Chem. Soc., 2007, 129, 2607 CrossRef CAS PubMed; K. Uemura, S. Kitagawa, K. Fukui and K. Saito, J. Am. Chem. Soc., 2004, 126, 3817 CrossRef PubMed; Y. Liu, R. Zhang, C. He, D. B. Dang and C. Y. Duan, Chem. Commun., 2010, 746 RSC.
- F. Larachi, J. Pierre, A. Adnot and A. Bernis, Appl. Surf. Sci., 2002, 195, 236 CrossRef CAS; P. Datta, P. Majewski and F. Aldinger, Mater. Charact., 2009, 60, 138 CrossRef.
- L. Zhao, Y. Liu, C. He, J. Wang and C. Y. Duan, Dalton Trans., 2014, 43, 335 RSC.
- K. J. Xu, J. Z. Zhao, D. Escudero, Z. Mahmood and D. Jacquemin, J. Phys. Chem. C, 2015, 119, 23801 CAS.
- I. K. Biernacka, A. Bartecki and K. Kurzak, Polyhedron, 2003, 22, 997 CrossRef.
- K. A. Connors, Binding Constants, John Wiley, New York, 1987 Search PubMed.
- G. Oster and Y. Nishijima, J. Am. Chem. Soc., 1956, 78, 1581 CrossRef CAS; B. Valeur, Molecular Fluorescence, Principles and Applications, Wiley-VCH Verlag GmbH, Weinheim, 2002 Search PubMed; J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, New York, 3rd edn, 2006 Search PubMed.
- A. B. Descalzo, K. Rurack, H. Weisshoff, R. Martinez-Manez, M. D. Marcos, P. Amoros, K. Hoffmann and J. Soto, J. Am. Chem. Soc., 2005, 127, 184 CrossRef CAS PubMed; F. Hibbert and J. Emsley, Adv. Phys. Org. Chem., 1990, 26, 255 CrossRef; C. He, Z. Lin, Z. He, C. Duan, C. Xu, Z. Wang and C. Yan, Angew. Chem., Int. Ed., 2008, 47, 877 CrossRef PubMed.
- C. He, J. Wang, P. Y. Wu, L. Y. Jia, Y. Bai, Z. C. Zhang and C. Y. Duan, Chem. Commun., 2012, 48, 11880 RSC.
- S. Neogi, M. K. Sharma and P. K. Bharadwaj, J. Mol. Catal. A: Chem., 2009, 299 Search PubMed; Y. Hwang, D.-Y. Hong, J. S. Chang, S. Jhung, Y.-K. Seo, J. Kim, A. Vimonet, M. Dautri, C. Serre and G. Ferey, Angew. Chem., Int. Ed., 2008, 47, 4144 CrossRef CAS PubMed.
- J. Zhang, H. Yu, C. X. Zhang, C. He and C. Y. Duan, New J. Chem., 2014, 38, 3137 RSC; Y. Jiao, J. Wang, P. Y. Wu, L. Zhao, C. He, J. Zhang and C. Y. Duan, Chem.–Eur. J., 2014, 20, 2224 CrossRef CAS PubMed.
- M. J. T. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. C. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. N. B. Mennucci, H. Petersson, M. Caricato, X. Li, H. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, Y. K. T. Nakajima, O. Honda, H. Nakai, T. Vreven, J. A. Montgomery Jr J. E. O. Peralta, F. Oligaro, M. Bearpark, J. J. Heyd, E. Brothers, V. N. Kudin, K. K. N. R. Staroverov, J. Normand, K. Raghavachari, A. B. Rendell, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Rega, M. Millam, J. E. Knox, J. B. Cross, V. Bakken, C. J. Adamo, J. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, J. W. Pomelli, J. W. Ochterski, R. L. Martin, K. Z. Morokuma, V. G. Zarkrzewski, G. A. Voth, P. Salvador, J. J. D. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision B.01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Characterization, relative luminescent and NMR spectra. See DOI: 10.1039/c5ra27806f |
| This journal is © The Royal Society of Chemistry 2016 |