
Thermodynamics of a food macromolecular assembly: the case of okra mucilage†
Abstract
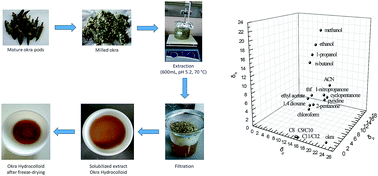
This is a comprehensive characterization of the bulk and interface thermodynamics of a model macromolecular hydrocolloid of interest in food, cosmetics and pharmaceutics, namely okra mucilage. Inverse gas chromatography (IGC) has been used to probe the affinity of 20 different compounds at infinite dilution to the hydrocolloid. Extensive surface characterization was conducted at three temperatures (40, 50, 60 °C) for the assessment of the surface energy, the free energy of adsorption, and the related enthalpic and entropic components. Okra extract matrix is shown to be Lewis amphoteric with predominantly basic character. Bulk thermodynamic parameters such as the Flory–Huggins interaction parameter, the weight fraction activity coefficient and the total and partial solubility parameters were determined at 90, 100, 110 °C. The above can form a background for the interpretation of many aspects of the hydrocolloid's structural and functional behavior.
 Please wait while we load your content...
Please wait while we load your content...

