
Smectic liquid crystal polymers as a template for ultrathin CaCO3 nanolayers†
Yifei Xuab,
Huub P. C. van Kuringencd,
Dirk J. Muldercd,
Albertus P. H. J. Schenning*ce and
Nico A. J. M. Sommerdijk*ae
aLaboratory of Materials and Interface Chemistry & Center of Multiscale Electron Microscopy, Department of Chemical Engineering and Chemistry, Eindhoven University of Technology, PO box 513, 5600 MB, Eindhoven, The Netherlands. E-mail: n.sommerdijk@tue.nl; Tel: +31 402475870
bNational Laboratory of Solid State Microstructures and School of Physics, Nanjing University, Hankou Rd.22, 210093, Nanjing, China. Fax: +86 25835925535; Tel: +86 2583592756
cLaboratory for Functional Materials and Devices, Department of Chemical Engineering and Chemistry, Eindhoven University of Technology, PO box 513, 5600 MB, Eindhoven, The Netherlands. E-mail: A.P.H.J.Schenning@tue.nl
dDutch Polymer Institute (DPI), P.O. Box 902, 5600 AX, Eindhoven, The Netherlands
eInstitute for Complex Molecular Systems, Eindhoven University of Technology, PO box 513, 5600 MB, Eindhoven, The Netherlands
First published on 27th January 2016
Abstract
The poly(aspartic acid) (pAsp) assisted infiltration of a nanoporous smectic liquid crystalline network with CaCO3 yields only ∼1 nm thick crystalline nanolayers. This bioinspired strategy opens the way to new functional materials based on solution-grown quasi 2D crystals of only a few unit cells thick.
In recent decades, numerous novel properties have been observed on ultrathin crystal nanolayers with quantum thickness (≤1 nm), such as the quantum Hall effect of graphene and giant magnetoresistance of Fe/Cr multilayers.1,2 These materials have promising application potential in data storage, catalysis, batteries and semiconductor devices.3–6 Up to now, ultrathin crystal nanolayers could only be synthesized by top-down methods such as magnetron sputtering (MS), molecular-beam-epitaxy (MBE) and laser-assisted catalyst growth (LCG).3,6,7 It is appealing to develop a low cost and efficient method to synthesize these nanolayers. A thermotropic smectic hydrogen bonded liquid crystal (LC)8,9 network with layered gaps of ∼1 nm thickness was fabricated in our previous study.10–14 The material provides an ideal template for infiltrating ultrathin crystal nanolayers. When the template was infiltrated by Ag ions, Ag nanoparticles rather than continuous sheets were prepared.12 This is mainly due to the Ag+ deficiency during the reaction. To fully infiltrate the polymer template with crystal nanolayers, solutes have to be continuously delivered to the gaps during the crystallization process.
Such a process is observed during the mineralization of collagen, e.g. in the formation of bone.15–17 Here collagen forms an organic biopolymer scaffold that is infiltrated with amorphous calcium phosphate which eventually crystallizes and deposits as aligned oriented platelets of apatite of only 2 nm in thickness. In fact these apatite platelets are the smallest crystals know in Nature.17
Poly-aspartic acid (pAsp) strongly binds to Ca2+ and significantly inhibits the crystallization of CaCO3 and calcium phosphate (CaP), stabilizing the otherwise unstable amorphous phases.18,19 By preventing crystallization pAsp promotes the infiltration of CaCO3 or CaP into the nanopores of synthetic membranes20,21 and has also been used to achieve the intrafibrillar mineralization of collagen with CaP in vitro.19,22,23 The success of this approach has been ascribed to the ability of pAsp, but also other charged polymers,24–28 to form a so-called polymer induced liquid precursor (PILP) phase which can infiltrate nanoporous templates through capillary action.18,19
Here we employ this bioinspired mineralization strategy to infiltrate a smectic LC porous network with CaCO3. In analogy to collagen mineralization it yields quasi-2D crystals with a thickness of only ∼1 nm, which depending on the polymorph and crystal orientation is only 3–4 unit cells thick. The method opens the way to the templated bottom-up synthesis of a wide range of quasi 2D materials of which the physical properties can be adjusted by the LC polymer network.
The experimental procedure is schematically illustrated in Fig. 1A. The smectic LC network was prepared using a previously reported method.12,13 Briefly: 4-(6-acryloyloxyl hexyloxy) benzoic acid (6OBA) and 4-((4-(6-(acryloyloxy) hexyloxy) phenoxy)carbonyl)phenyl 4-(6-(acryloyloxy) hexyloxy) (C6H) (structures shown in Fig. 1B) were mixed in a 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio to form a smectic LC phase. This phase was photo-polymerized under UV light, resulting in a free standing smectic polymer network of approximately 0.5 cm × 0.5 cm × 18 μm in size. The film was then treated in 0.05 M KOH solution to break up the H-bonds. The benzoate groups become randomly oriented while the C6H remain unaxially oriented, resulting in layered gaps with a pore size of ∼1 nm.13 This led to swelling and a loss of the order within the layer and a concomitant loss of the birefringence in polarized optical microscopy (POM).13 The KOH treated membrane was washed with de-ionized water and then immersed overnight in a 10 mM CaCl2 solution containing 25 mg l−1 or 100 mg l−1 pAsp at room-temperature. The pH value of this solution was adjusted to pH 9 using 0.1 M KOH solution to keep the gaps open.13
:1 ratio to form a smectic LC phase. This phase was photo-polymerized under UV light, resulting in a free standing smectic polymer network of approximately 0.5 cm × 0.5 cm × 18 μm in size. The film was then treated in 0.05 M KOH solution to break up the H-bonds. The benzoate groups become randomly oriented while the C6H remain unaxially oriented, resulting in layered gaps with a pore size of ∼1 nm.13 This led to swelling and a loss of the order within the layer and a concomitant loss of the birefringence in polarized optical microscopy (POM).13 The KOH treated membrane was washed with de-ionized water and then immersed overnight in a 10 mM CaCl2 solution containing 25 mg l−1 or 100 mg l−1 pAsp at room-temperature. The pH value of this solution was adjusted to pH 9 using 0.1 M KOH solution to keep the gaps open.13
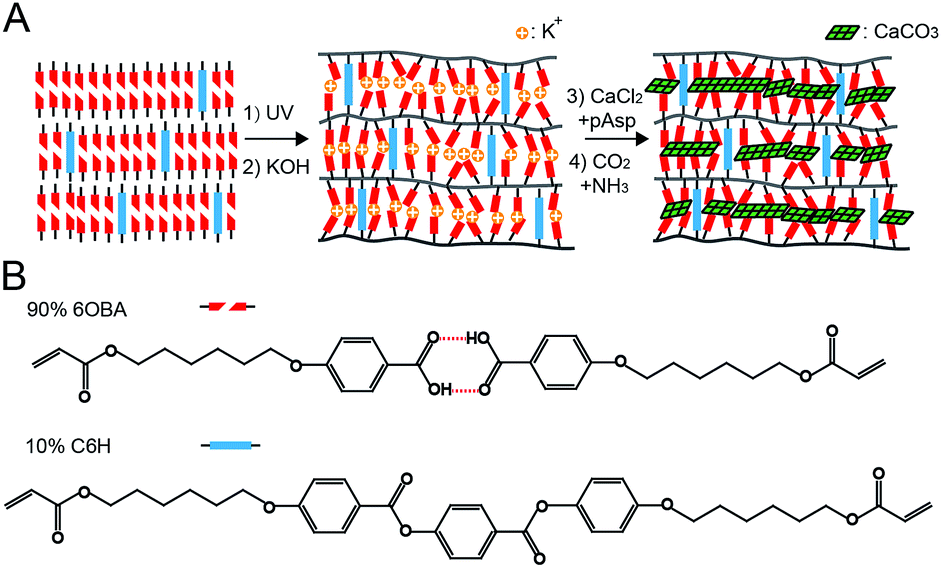 | ||
| Fig. 1 (A) Schematic representation of the experimental process: the smectic LC of a 6OBA/C6H mixture is photo-polymerized by UV light to form a nanoporous polymer network upon treatment with KOH. The membrane is then immersed in a CaCl2/pAsp solution and exposed to a mixture of CO2/NH3 through (NH4)2CO3 decomposition. Eventually crystalline CaCO3 nanolayers are formed within the nanogaps. (B) The chemical structures of the 6OBA and C6H, respectively. | ||
The formation of ultrathin CaCO3 nanolayers within the layered gaps was approached by allowing a CO2/NH3 mixture gas to diffuse into the solution using the decomposition of solid (NH4)CO3.29 In the solution the CO2 converted into carbonate ions which reacted with the Ca2+ ions to form CaCO3 on/within the membrane, while the influx of NH3 kept the pH value constant at ∼10 to both support the CaCO3 formation and prevent the recovery of H-bonds.
POM of the membrane after 24 h of CaCO3 growth in the presence of 25 mg l−1 pAsp (Fig. 2A) showed that the membrane was covered by micron sized CaCO3 particles while the re-appearance of the birefringence suggested an increased structural order. Comparison of the Fourier transform infra-red (FTIR) spectra before and after mineralization revealed an extra peak at 1675 cm−1, corresponding to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O of carboxyl groups in their hydrogen bonded state, indicating that part of the H-bonds had been recovered (Fig. 2B).13 At the same time, the carbonate ν3 peak in CaCO3 appeared at 1450 cm−1, while the carboxylate νs peak at 1546 cm−1 shifted to lower wavenumber, indicating complexation of Ca ions and providing evidence that CaCO3 crystals were formed not only on the surface, but also within the membrane.13,30,31
O of carboxyl groups in their hydrogen bonded state, indicating that part of the H-bonds had been recovered (Fig. 2B).13 At the same time, the carbonate ν3 peak in CaCO3 appeared at 1450 cm−1, while the carboxylate νs peak at 1546 cm−1 shifted to lower wavenumber, indicating complexation of Ca ions and providing evidence that CaCO3 crystals were formed not only on the surface, but also within the membrane.13,30,31
 | ||
| Fig. 2 Analysis of the sample grown in the presence of 25 mg l−1 pAsp for 24 h (A) POM image. Dark areas point to the formation of CaCO3 crystals. Polarizer directions are indicated by white arrows (B) FTIR spectrum of the film. The spectrum of membrane after KOH treatment is shown for comparison. (C–E) TEM images of a microtomed thin section. (C) Low magnification image. Two areas 1 and 2 are highlighted by red dash line circles. The insets show SAED patterns of area 1 and 2, respectively. Spots corresponding to calcite or vaterite diffractions are indexed by “C” or “V”, respectively. (D) Enlargement of area 2. The inset shows the FFT image of the area within the red dash line square. Two spots centered at d = 5.0 and 2.5 nm are highlighted by yellow arrows, respectively. (E) High-resolution/near focus image of (D), the separation distance D1 and layer thickness D2 and are indicated by yellow lines. The inset shows the FFT of the image. A pair of spots at d = 3.3 Å are highlighted by yellow dash line circles. | ||
Ultramicrotomy was used to prepare thin sections of the membranes which were subsequently examined by transmission electron microscopy (TEM). It should be noted that the membrane became rigid and curled up after CaCO3 mineralization, making the actual cutting angle difficult to be controlled. Investigation of the membrane cross sections showed the mineralized membrane surface (ESI, Fig. S1†), of which parts were fully occupied by CaCO3 (Fig. 2C, area 1). Selected area electron diffraction (SAED) demonstrated that CaCO3 was present in two polymorphic types: calcite and vaterite (calcite: JCPDF #72-1937, vaterite: JCPDF #72-0506) (Fig. 2C, inset 1) that showed multiple crystallographic orientations.
Other areas showed less contrast indicating a lower degree of mineralization (Fig. 2C, area 2). Indeed, the SAED patterns of these regions showed fewer spots, which also in this case pointed to a mixture of calcite and vaterite (Fig. 2C, inset 2). Higher magnification images of this area showed that they were composed of bent, but well-aligned ultrathin nanolayers of the mineral embedded in the template (Fig. 2E). FFT analysis image (Fig. 2D, inset) revealed a pair of spots representing a d-value of 5.0 nm in the direction perpendicular to the layers, with a deviation of ∼0.5 nm, indicating a uniform separation distance for the CaCO3 nanolayers. Additional weaker spots with d-value of ∼2.5 nm in the same direction were attributed to secondary FFT signals. Direct measurements on high-resolution/near-focus TEM images of this area (Fig. 2E) confirmed a separation distance (D1) of 5.0 nm, and further revealed a layer thickness (D2) of ∼1.7 nm. It should, however, be noted that these values depend on the angle the cross section makes with the layers, and that all angles that deviate from a perfect perpendicular cut will increase the observed distances. Since the actual separation distance of the layers was previously determined to be ∼3.7 nm at high humidity,13 we can correct for this angular deviation and determine the actual CaCO3 layer thickness to be ∼1.3 nm, in line with the previous results. Although the sample thickness prevented us to obtain high resolution TEM images with lattice resolution (Fig. 2E), the FFT showed a pair of weak spots with d = 3.3 Å that may be attributed to the (011) plane of vaterite (Fig. 2E, inset).
The results showed that the membrane was readily infiltrated by CaCO3 crystal nanolayers. In addition we found that no infiltration occurred in regions where large CaCO3 crystals had nucleated on the outside of the membrane (ESI, Fig. S2†). Probably, the formation of crystals reduced the local supersaturation and the crystals acted as a sink for the ions present in the solution and prohibit the diffusion of the mineral into the film. Continued exposure to the mineralization solution for 7 days resulted in the complete mineralization of the membrane with a mixture of calcite and vaterite (ESI, Fig. S3†) but also in the deposition of more large crystals on the membrane surface. Surprisingly the increase in the intensity of the 1675 cm−1 vibration in FTIR suggested that crystallization in the nanogaps goes hand-in-hand with the reformation of the H-bonds, which must be at the expense of the interaction between the carboxylates and the mineral.
To reduce precipitation of CaCO3 on the membrane surface, the pAsp concentration [pAsp] was increased to 100 mg l−1. After 24 h of mineralization POM indeed detected less CaCO3 particles on the surface (Fig. 3A). In line with this, FTIR spectrum (Fig. 3B) showed no H-bond signal at 1675 cm−1, and also the CaCO3 peak at 1450 cm−1 was weaker compared with the sample prepared at a [pAsp] = 25 mg l−1. Also for this sample TEM of a microtomed thin cross section showed high contrast bands pointing at significant mineralization (Fig. 3C). However, SAED showed broad rings indicating the mineral is still amorphous (Fig. 3C, inset) and suggesting the area is occupied by amorphous calcium carbonate (ACC) even though overlapping signals prevented confirmation with FTIR (Fig. 3B). Higher magnification imaging showed that the ACC forms nanolayers (Fig. 3D). FFT of the TEM images again showed 3 pairs of spots in the direction perpendicular to the nanolayers with d-values of 6.3 ± 0.6, 3.2 ± 0.2 and 2.1 ± 0.1 nm, respectively (inset of Fig. 3D), which should belong to the same diffraction group. This indicated the ACC nanolayers also have a uniformed separation distance and thickness. Direct measurements of the features in the TEM images revealed a separation distance (D1) of 6.3 nm which is in line with the d-value of the primary FFT signal, and a layer thickness (D2) of ∼2.0 nm. Again taking into account a likely non-perpendicular angle of the thin section with the direction of the layers, the actual thickness of the mineral layers here should be ∼1.2 nm, hence similar to the values determined for the crystalline layers obtained at lower [pAsp] (Fig. 2). The results showed that at higher [pAsp], the CaCO3 crystallization was further inhibited, with the nanolayers maintaining the amorphous state and no reestablishment of H-bonds within 24 h.
 | ||
| Fig. 3 Analysis of the sample grown in the presence of 100 mg l−1 pAsp for 24 h (A) POM image. Dark areas point to the formation of CaCO3 crystals. Polarizer directions are indicated by white arrows (B) FTIR spectrum of the film. The spectrum of 25 mg l−1 pAsp, 24 h sample is used for comparison. (C and D) TEM images of a microtomed thin section. (C) Low magnification image. The inset shows the SAED pattern of the area within the red dash line circle. (D) Higher magnification of (C) showing the nanolayers. The separation distance D1 and the layer thickness D2 and are schematically illustrated by yellow lines. The inset shows the FFT of the image. Three spots centered at d = 6.3, 3.2 and 2.1 nm are highlighted, respectively. | ||
Based on previous studies18,20 we may assume that also at [pAsp] = 25 mg l−1 the mineral first deposits as an amorphous phase, but that at this lower polymer concentration the amorphous-to-crystal transition occurs within the 24 h reaction period. It is interesting to note that this transformation was delayed when increasing polymer concentration, similar to what was observed for the infiltration of collagen with CaP.32 As the mineral was formed under identical reaction conditions and in the same templating membrane, this implies that it is the polymer that is responsible for this delay. This will allow using these polymers also to further fine tune the structure and properties of the inorganic nanolayers. We have previously shown that although pAsp aids in the intrafibrillar mineralization of collagen, this process does not require the polymer to enter the collagen fibril.22 Our present results raise the question to if pAsp is able to penetrate the ultra-small gaps of the membrane, and to what extend this related to the observed delay in crystallization. It is intriguing to speculate that a similar mechanism could be operational during the mineralization of collagen.
In summary, we have successfully infiltrated a polymerized smectic LC network membrane with ∼1 nm thick CaCO3 nanolayers, using a bio-inspired approach analogous to the mineralization of collagen. Using pAsp as a crystallization control agent that inhibits immediate precipitation, an amorphous mineral precursor was slowly and continuously delivered to the nanogaps, allowing the formation of ultrathin quasi-2D crystals of only 3–4 unit cell thick. This method can in principle be extended to a wide variety of crystalline materials that can be grown from solution at mild temperatures and at pH > 9, such as ZnO or Fe3O4.33,34 By using different cross-linkers, the thickness and separation distances of the nanolayers could be further tuned depending on the application requirement.14 This work therefore provides a promising generic pathway for the bottom up synthesis of ultrathin crystal nanolayers.
Acknowledgements
YX is financially supported by the China Scholarship Council (CSC). The work of NAJMS is supported by a VICI grant from the Netherlands Organization of Scientific Research. This research of HvK and DJM forms part of the research programme of the Dutch Polymer Institute (DPI), project 742 & 776.Notes and references
- Y. Zhang, Y. W. Tan, H. L. Stormer and P. Kim, Nature, 2005, 438, 201–204 CrossRef CAS PubMed.
- M. N. Baibich, J. M. Broto, A. Fert, F. N. Van Dau, F. Petroff, P. Etienne, G. Creuzet, A. Friederich and J. Chazelas, Phys. Rev. Lett., 1988, 61, 2472–2475 CrossRef CAS PubMed.
- C. Chappert, A. Fert and F. N. Van Dau, Nat. Mater., 2007, 6, 813–823 CrossRef CAS PubMed.
- L. Qu, Y. Liu, J. B. Baek and L. Dai, ACS Nano, 2010, 4, 1321–1326 CrossRef CAS PubMed.
- E. Yoo, J. Kim, E. Hosono, H. S. Zhou, T. Kudo and I. Honma, Nano Lett., 2008, 8, 2277–2282 CrossRef CAS PubMed.
- X. Duan, Y. Huang, Y. Cui, J. Wang and C. M. Lieber, Nature, 2001, 409, 66–69 CrossRef CAS PubMed.
- S. Yuasa, A. Fukushima, H. Kubota, Y. Suzuki and K. Ando, Appl. Phys. Lett., 2006, 89, 042505 CrossRef.
- T. Kato, H. Kihara, U. Kumar, T. Uryu and J. M. J. Fréchet, Angew. Chem., Int. Ed. Engl., 1994, 33, 1644–1645 CrossRef.
- H. Kihara, T. Kato, T. Uryu and J. M. J. Fréchet, Chem. Mater., 1996, 8, 961–968 CrossRef CAS.
- C. L. Gonzalez, C. W. M. Bastiaansen, J. Lub, J. Loos, K. Lu, H. J. Wondergem and D. J. Broer, Adv. Mater., 2008, 20, 1246–1252 CrossRef CAS.
- I. K. Shishmanova, C. W. Bastiaansen, A. P. H. J. Schenning and D. J. Broer, Chem. Commun., 2012, 48, 4555–4557 RSC.
- D. Dasgupta, I. K. Shishmanova, A. Ruiz-Carretero, K. Lu, M. Verhoeven, H. P. C. van Kuringen, G. Portale, P. Leclère, C. W. M. Bastiaansen and D. J. Broer, J. Am. Chem. Soc., 2013, 135, 10922–10925 CrossRef CAS PubMed.
- H. P. C. van Kuringen, G. M. Eikelboom, I. K. Shishmanova, D. J. Broer and A. P. H. J. Schenning, Adv. Funct. Mater., 2014, 24, 5045–5051 CrossRef CAS.
- H. P. C. van Kuringen, Z. J. W. A. Leijten, A. H. Gelebart, D. J. Mulder, G. Portale, D. J. Broer and A. P. H. J. Schenning, Macromolecules, 2015, 48, 4073–4080 CrossRef CAS.
- W. Traub, T. Arad and S. Weiner, Proc. Natl. Acad. Sci. U. S. A., 1989, 86, 9822–9826 CrossRef CAS.
- D. J. Hulmes, T. J. Wess, D. J. Prockop and P. Fratzl, Biophys. J., 1995, 68, 1661–1670 CrossRef CAS PubMed.
- L. C. Palmer, C. J. Newcomb, S. R. Kaltz, E. D. Spoerke and S. I. Stupp, Chem. Rev., 2008, 108, 4754–4783 CrossRef CAS PubMed.
- L. B. Gower, Chem. Rev., 2008, 108, 4551–4627 CrossRef CAS PubMed.
- M. J. Olszta, X. Cheng, S. S. Jee, R. Kumar, Y. Y. Kim, M. J. Kaufman, E. P. Douglas and L. B. Gower, Mater. Sci. Eng., R, 2007, 58, 77–116 CrossRef.
- Y. Y. Kim, N. B. Hetherington, E. H. Noel, R. Kröger, J. M. Charnock, H. K. Christenson and F. C. Meldrum, Angew. Chem., 2011, 123, 12780–12785 CrossRef.
- B. Cantaert, E. Beniash and F. C. Meldrum, J. Mater. Chem. B, 2013, 1, 6586–6595 RSC.
- F. Nudelman, K. Pieterse, A. George, P. H. Bomans, H. Friedrich, L. J. Brylka, P. A. Hilbers, G. de With and N. A. J. M. Sommerdijk, Nat. Mater., 2010, 9, 1004–1009 CrossRef CAS PubMed.
- A. S. Deshpande and E. Beniash, Cryst. Growth Des., 2008, 8, 3084–3090 CAS.
- T. Kato, Adv. Mater., 2000, 12, 1543–1546 CrossRef CAS.
- N. A. J. M. Sommerdijk, E. N. M. van Leeuwen, M. R. J. Vos and J. A. Jansen, CrystEngComm, 2007, 9, 1209–1214 RSC.
- B. Cantaert, Y. Y. Kim, H. Ludwig, F. Nudelman, N. A. J. M. Sommerdijk and F. C. Meldrum, Adv. Funct. Mater., 2012, 22, 907–915 CrossRef CAS.
- A. S. Schenk, H. Zope, Y. Y. Kim, A. Kros, N. A. J. M. Sommerdijk and F. C. Meldrum, Faraday Discuss., 2012, 159, 327–344 RSC.
- T. Nishimura, K. Toyoda, T. Ito, Y. Oaki, Y. Namatame and T. Kato, Chem.–Asian J., 2015, 10, 2356–2360 CrossRef CAS PubMed.
- L. Addadi, J. Moradian, E. Shay, N. G. Maroudas and S. Weiner, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 2732–2736 CrossRef CAS.
- N. Vagenas, A. Gatsouli and C. Kontoyannis, Talanta, 2003, 59, 831–836 CrossRef CAS PubMed.
- M. Moirangthem, R. Arts, M. Merkx and A. P. H. J. Schenning, Adv. Funct. Mater., 2016 DOI:10.1002/adfm.201504534.
- F. Nudelman, P. H. H. Bomans, A. George, G. de With and N. A. J. M. Sommerdijk, Faraday Discuss., 2012, 159, 357–370 RSC.
- Z. Liu, X. Wen, X. Wu, Y. Gao, H. Chen, J. Zhu and P. Chu, J. Am. Chem. Soc., 2009, 131, 9405–9412 CrossRef CAS PubMed.
- J. J. M. Lenders, H. R. Zope, A. Yamagishi, P. H. H. Bomans, A. Arakaki, A. Kros, G. de With and N. A. J. M. Sommerdijk, Adv. Funct. Mater., 2015, 25, 711–719 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, TEM images of the mineralized membrane, the regions covered by CaCO3 particles and experimental results of the sample grown for 7 days. See DOI: 10.1039/c5ra27752c |
| This journal is © The Royal Society of Chemistry 2016 |