
A two-step continuous synthesis of α-ketoamides and α-amino ketones from 2° benzylic alcohols using hydrogen peroxide as an economic and benign oxidant†
Kai
Guo  *ac
*ac
*ac
Abstract
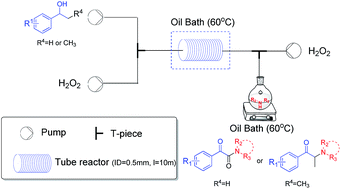
A practical two-step synthesis of α-ketoamides and α-amino ketones via direct oxidative coupling between 2° benzylic alcohols and amines was developed. Hydrogen peroxide, an economic and environmentally friendly oxidant, was used, and a metal catalyst was unnecessary. Moreover, the continuous-flow technique was employed to increase the functional group tolerance, efficiency and safety.
 Please wait while we load your content...
Please wait while we load your content...

