
Self-induced organic guest packed in three-dimensional architecture based on hetero-alkali metallic sulfonatothiacalix[4]arene†
Uma Maheswara Rao Kundaa,
Manabu Yamadab,
Hiroshi Katagiric and
Fumio Hamada*d
aVenture Business Laboratory, Akita University, 1-1 Tegatagakuen-machi, Akita, 010-8502, Japan
bResearch Center for Engineering Science, Graduate School of Engineering and Resource Science, Akita University, 1-1 Tegatagakuen-machi, Akita, 010-8502, Japan
cGraduate School of Science and Engineering, Yamagata University, 4-3-16 Jonan, Yonezawa, Yamagata 992-8510, Japan
dDepartment of Applied Chemistry for Environments, Graduate School of Engineering and Resource Science, Akita University, 1-1 Tegatagakuen-machi, Akita, 010-8502, Japan. E-mail: hamada@gipc.akita-u.ac.jp; Fax: +81 18 889 2714; Tel: +81 18 889 2714
First published on 29th January 2016
Abstract
The inclusion behavior of pyridine N-oxide in the cavity of hetero-alkalimetallic (Na, K) sulfonatothiacalix[4]arene was studied by single crystal X-ray diffraction. This hetero-alkali metallic host–guest assembly (3) was mainly supported by π–π stacking, S–π interactions and hydrogen bonding. In addition, all metal ions significantly contributed for stabilizing the architecture via coordination with host and guest molecules. We also studied the thermogravimetric analysis, dipole moment and energy gap between frontier molecular orbitals of 3.
Introduction
Understanding the nature and energetics of non-covalent interactions through X-ray crystal structures that govern the design of new solids with desired physical and chemical properties is the aim of crystal engineering. Due to the remarkable inclusion and metal coordination abilities, calixarenes and thiacalixarenes are contributing significantly to this field.1 The presence of hydrophobic cavities in these molecules arising from the phenyl rings allows the binding of various guests with specific non-covalent interactions. Sulfur bridges instead of methylene bridges are the advantage of thiacalixarenes over calixarenes, which extended their coordination ability to the lower rim along with creating a wider cavity and more flexibility.2 Thus, thiacalixarenes, particularly water soluble thiacalixarenes, have received considerable attention because their appearance in the chemical sciences.3The inclusion properties and metal coordination of p-sulfonatothiacalix[4]arene (1) were studied by various researchers around the world, and these reports reveal the huge differences in coordination behavior of 1 over the water soluble calix[4]arene, even though their shapes and structures were similar.4 These complexes were mostly stabilized in the cone conformation of the 1 unit with up-down arrangement of bilayer structure. However, some exceptions from the cone conformation were observed in the inclusion complexes with 1,4-dioxane, 2,2′-bipyridine, and 4,4′-bipyridine. Compound 1 adopts a 1,2-alternate conformation with 1,4-dioxane and 2,2′-bipyridine, whereas it adopts a partial cone conformation with 4,4′-bipyridine.5 These findings reveal that the guest molecule influences the conformation of 1 due to high flexibility of the cavity. Inclusion of small organic guest molecules allows 1 to adopt the original cone shape with C4v symmetry, and pinched cones with C2v symmetry were observed in the case of large guest molecules.6
The complexation of 1 with Cu(NO3)2 and pyridine-N-oxide (PNO) results in a dimeric capsule-like supramolecular architecture.7 The PNO molecule occupies the cavity of 1 and is coordinated to Cu ions, and additional Cu ions are ligated to the sulfonate oxygen of 1. In this complex, Cu acts as a bridge between the host and guest molecules. The inclusion of PNO in 1 in the presence of trivalent lanthanide ions (Ln(III), Ln = La, Nd) was described recently by Guo Q et al.8 In addition to the same structural features as that with copper complex of 1 and PNO, Ln(III) ions were also coordinated to the bridged sulfur and two phenolic oxygens. Different phenomena were observed in the case of water soluble calix[4]arene complexation with trivalent lanthanide ions and PNO. Inclusion behavior of PNO was dependent on its molar ratio, which results in two different inclusion complexes that were described as spherical and tubular assemblies by J. L. Atwood et al.9 In addition to lanthanides, sodium ions are also involved in stabilizing these supramolecular capsule assemblies.
The above discussion reveals that the inclusion of PNO in a water-soluble calixarene and thiacalixarene resulted in interesting supramolecular assemblies with numerous non-covalent interactions, which play a significant role in structural chemistry. However, this study was limited to lanthanides and transition metals in both calixarene and thiacalixarene chemistry. We extended this study towards alkali metals; a hetero-alkali metallic supramolecular assembly of 1 with PNO occupying its cavity is reported in the present study, and interesting structural features were observed in this complex. To the best of our knowledge, this is the first example of self-induced aromatic guest complexation of hetero-alkali metallic three-dimensional supramolecular assembly in thiacalixarenes.
Results and discussion
In our previous study, we reported a hetero-alkali metallic (Na, K) supramolecular assembly based on 1.10 The complex Na2K4[p-sulfonatothiacalix[4]arene]·7H2O (2) was stabilized with a variety of interesting non-covalent interactions such as aromatic π hydrogen bonding, π–π stacking, and S–π interactions. Furthermore, the hydrated metal ions were coordinated with the bridged sulfur, phenolic oxygens and sulfonate oxygens with various coordination numbers. The water molecules played a significant role in stabilizing this bilayer assembly through hydrogen bonding.In addition, we studied the inclusion properties of 1 in the presence of K2CO3 with PNO. For this purpose, K2CO3 and PNO were added to the aqueous solution of 1. The mixture was allowed to slowly evaporate at room temperature. After several days, crystals suitable for single crystal X-ray diffraction studies were formed. 1H-NMR data reveals that the shifting of the aromatic protons of PNO towards higher field from that of the independent PNO molecule is due to its inclusion in hydrophobic cavity of 1. The three signals of the PNO molecule, that is, the doublet, doublet of doublet and triplet, shifted from δ8.34, δ7.80 and δ7.64 to δ8.20, δ7.45, and δ7.38, respectively.
The single crystal X-ray diffraction analysis reveals that the complex was stabilized via fascinating non-covalent interactions. The asymmetric unit of Na2K4[p-sulfonatothiacalix[4]arene (PNO)]·7H2O (3) was comprised of one molecule of 1, one molecule of PNO, four K ions, two Na ions and seven water molecules. The PNO molecule was imbedded in the hydrophobic cavity of 1 and further linked to three K ions, namely, K2, K3 and K4 through the oxide ion. Unlike that in the other PNO inclusion complexes in the calixarene family, the oxide ion adopts a tetrahedral geometry. The copper complex with PNO and 1, and Ln complex with PNO and 1 (Ln = La, Nd), showed clear distortion from C4v to C2v symmetry of 1 due to the inclusion of the PNO molecule in the cavity. The S⋯S distances of the trans sulfonate groups (10.430 Å, 9.503 Å) indicate that complex 3 maintains the C4v symmetry. The four phenyl rings of the one unit of the assembly are designated as A, B, C and D (Fig. 1).
 | ||
| Fig. 1 Asymmetric unit of 2. A, B, C and D represent C1–C6, C7–C12, C13–C18 and C19–C24, respectively. Each atom is depicted as follows: K = violet, Na = light blue; S = yellow; O = red; N = light violet; and C = gray. Hydrogens were omitted for clarity. | ||
The imbedded PNO molecule exhibits non-classical hydrogen bonding with the centroids of the aromatic rings with distance 3.586 Å (C26⋯centroid C) and 2.976 Å (CH26⋯centroid C) (Fig. S1†). Though non-classical hydrogen bonding is weaker than classical hydrogen bonding, it has significant importance in biological systems.11 Two water molecules were imbedded in the cavity of 1 in complex 2 and exhibited aromatic π hydrogen bonding. However, in this complex, these two water molecules changed their direction in the presence of PNO and settled outside the cavity. Furthermore, the asymmetric unit was stabilized by hydrogen bonding between water molecules and sulfonate oxygens with distances 2.800 Å (O5⋯O5W), 2.004 Å (O5⋯H5W2), 3.036 Å (O11⋯O6W), and 2.640 Å (O11⋯H6W) (Fig. S1†). The distances between the aromatic carbons of PNO and surrounding potassium ions (3.915 Å (K1⋯C28), 3.890 Å (K3⋯C25), 3.715 Å (K4⋯C29)) are less than the sum of their van der Waals radii (4.45 Å), which implies strong packing of the host–guest complex.
The extended structure of 3 reveals that all the phenyl moieties were held together by π–π aromatic stacking interactions with antiparallel aromatic rings, resulting in the three-dimensional supramolecular assembly. In complex 2, four different distances were observed between the stacked phenyl rings, but in complex 3, centroid A interacts with centroid Da (3.745 Å) and centroid B interacts with centroid Cb (3.888 Å) (Fig. 2). The symmetry elements are a1 − x, −1/2 + y, 1/2 − z, and b2 − x, 1/2 + y, 1/2 − z. In addition, the assembly was further supported by S–π interactions12 observed between the bridged sulfur and neighboring aromatic rings (Fig. 2). The corresponding distances and angles are 3.761 Å (S2⋯centroid Da), 162.49° (α, C8⋯S2⋯centroid Da), 64.55° (α′, C6⋯S2⋯centroid Da), 63.44° (φ, S2⋯centroid Da⋯C24a) (Fig. 3a), 3.757 Å (S4⋯centroid Bb), 166.74° (α, C20⋯S4⋯centroid Bb), 67.02° (α′, C18⋯S4⋯centroid Bb), and 62.87° (φ, S4⋯centroid Bb⋯C12b) (Fig. 3b). The symmetry elements are a1 − x, −1/2 + y, 1/2 + y, and b2 − x, 1/2 + y, 1/2 − z.
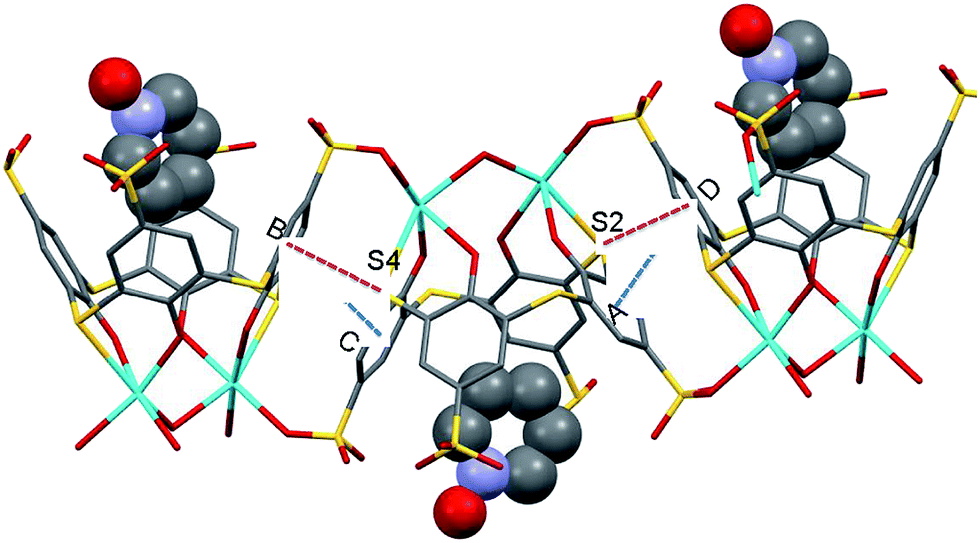 | ||
| Fig. 2 Extended structure of 3 showing π⋯π stacking (blue dotted lines) and S⋯π interactions (red dotted lines). PNO molecule is represented in spacefill model. Hydrogens were omitted for clarity. Each atom is depicted as follows: Na = light blue; S = yellow; O = red; N = light violet; and C = gray. | ||
 | ||
| Fig. 3 (a) S⋯π interaction between bridged sulfur (S2) and phenyl ring (Da). (b) S⋯π interaction between bridged sulfur (S4) and phenyl ring (Bb). Each atom is depicted as follows: S = yellow; O = red; C = gray; H = white; and Na = light blue. | ||
Furthermore, the PNO included sulfonatothiacalix[4]arene units of the overall assembly of 3 were arranged horizontally in ‘up-down’ and vertically in ‘pole-like’ fashion with the same directional units when viewed along with ‘yz’ plane (Fig. 4), resulting in two different directional layers. These layers were stabilized by π–π aromatic stacking and S–π interactions. Though up-down arrangement was observed in the case of 2, the vertical pole-like arrangement was not observed. This difference might be due to absence of lower rim coordination of the K ion with the bridged sulfur and phenolic oxygen in 3.
 | ||
| Fig. 4 Overall crystal structure of 3 showing horizontal ‘up-down’ and vertical ‘pole-like’ alternating layers when viewed from ‘yz’ plane. | ||
The S⋯S distances between bridged sulfur atoms of neighboring 1 units in 2 is greater than the sum of their van der Waals radii (ranging from 3.602 Å to 4.392 Å). However, in this case, distances less than the sum of their van der Waals distances (3.525 Å (S⋯S1b) and 3.479 Å (S3⋯S4c)) were observed. This probably represents the influence of an organic guest molecule on the packing of the supramolecular assembly by its interaction with the host molecule. Furthermore, the hydrogen bonding between neighboring units among water molecules, between water molecules and bridged sulfur atoms and between water molecules and sulfonate oxygens make the assembly a three-dimensional hydrogen bonded architecture.
The inclusion of PNO largely influenced the metal coordination when comparing complex 3 with complex 2. In complex 2, all metal ions are stabilized by various coordination numbers; moreover, one K and two Na ions coordinated to bridged sulfur and phenolic oxygen through lower rim coordination. In the case of complex 3, all K ions were stabilized with 8 coordination number without lower rim coordination and Na ions were stabilized with 6 coordination with similar ligands to those in complex 2. All metal ions act as bridges between 1 units to construct the three dimensional supramolecular assembly. Each Na coordinates two phenolic oxygens, one bridged sulfur of the asymmetric unit and two sulfonate oxygens of neighboring units (Fig. S2†). K1 coordinates with two sulfonate oxygens and one water molecule of the asymmetric unit and three sulfonate oxygens and two water molecules of neighboring units (Fig. S3†); K2 and K4 each coordinate with one sulfonate oxygen, the oxygen of PNO and two water molecules of the asymmetric unit and three sulfonate oxygens and one water molecule of neighboring units (Fig. S4 and S6†), whereas K3 coordinates with two sulfonate oxygens, the oxygen of PNO and one water molecule of the asymmetric unit and four sulfonate oxygens of neighboring units (Fig. S5†).
Thermogravimetric analysis data represents the stability of 3 (Fig. 5), and it indicates the gradual loss of water molecules from 70.43 °C. The weight loss at 185 °C (ca. 9.32%, calcd 9.22%) corresponds to escape of all water molecules from the complex. Further significant decrease of weight loss was observed 333 °C onwards due to the decomposition of the organic compound.
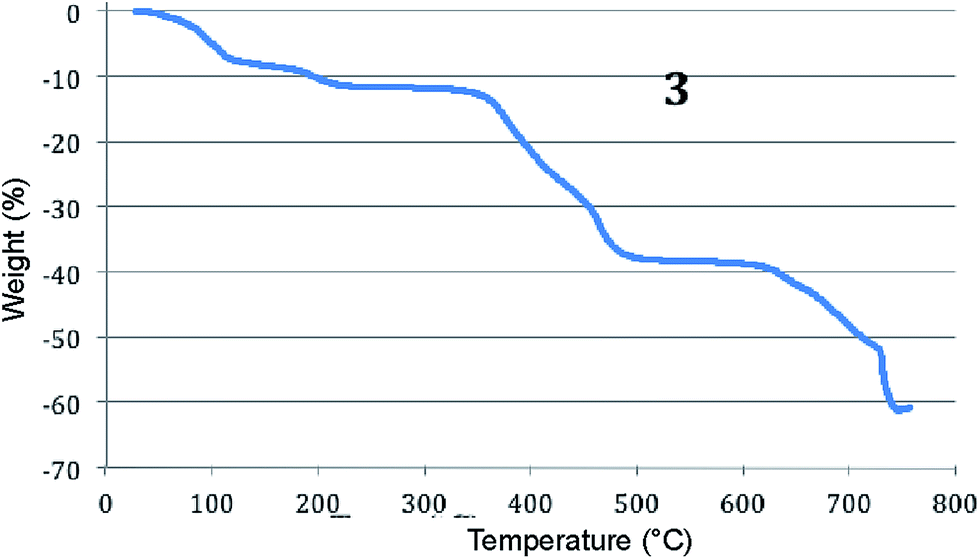 | ||
| Fig. 5 Thermogravimetric analysis of 3 recorded in the temperature range between 30 and 800 °C at a heating rate of 10 °C min−1. | ||
We also studied the energy gap between frontier molecular orbitals of 3 using Spartan-10 software and compared it with that of 2 (Fig. S7†). The major difference between these two complexes is the guest molecules in their respective cavities. The cavity of 3 was occupied with a PNO molecule, whereas the cavity of 2 was occupied with two water molecules. Though the energy gaps between the frontier molecular orbitals of both complexes were close to each other, the dipole moment was largely affected by the inclusion of PNO. The dipole moments for 2 and 3 were 71.18 debye and 17.85 debye, respectively. The massive decrease in the polarity of 3 might be due to opposite orientation of host and guest molecules.
Experimental
Synthesis and characterization of 3 and 4
X-ray crystallography
The crystals in mother liquid were picked up with a pipette and dropped in Paratone. The single crystals coated with oil were isolated on MicromountsTM, and the crystals were placed in a cold nitrogen stream at 93 K. X-ray diffraction data were collected on a Rigaku Saturn CCD diffractometer equipped with graphite-monochromated Mo Kα radiation. The structure was solved by direct methods using SHELXS-97 and refined using the full-matrix least-squares method on F2 using the SHELXL-2014 program.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 784 measured reflections, 10140 unique reflections (Rint = 0.0509), 9170 observed reflections, 671 parameters, R = 0.0444(I > 2.00σ(I)), wR = 0.0955 (all data), refined against [F], GOF = 1.123.
784 measured reflections, 10140 unique reflections (Rint = 0.0509), 9170 observed reflections, 671 parameters, R = 0.0444(I > 2.00σ(I)), wR = 0.0955 (all data), refined against [F], GOF = 1.123.Conclusions
In conclusion, we have demonstrated the inclusion behavior of PNO towards 1 in the presence of K2CO3. It reveals the formation of a hetero-alkali metallic supramolecular assembly in which the hydrophobic cavity was occupied by the PNO molecule. The guest molecule was further supported by coordination of three potassium ions with its oxygen ion. The overall supramolecular assembly was stabilized by metal coordination, π–π stacking, S–π interactions and hydrogen bonding.References
- (a) R. Kumar, Y. O. Lee, V. Bhalla, M. Kumar and J. S. Kim, Chem. Soc. Rev., 2014, 43, 4824 RSC; (b) N. Morohashi, F. Narumi, N. Iki, T. Hattori and S. Miyano, Chem. Rev., 2006, 106, 5291 CrossRef CAS PubMed; (c) P. Jin, S. J. Dalgarno and J. L. Atwood, Coord. Chem. Rev., 2010, 254, 1760 CrossRef CAS.
- (a) N. Iki and S. Miyano, J. Inclusion Phenom. Macrocyclic Chem., 2001, 41, 99 CrossRef CAS; (b) N. Morohashi, S. Noji, H. Nakayama, Y. Kudo, S. Tanaka, C. Kabuto and T. Hattori, Org. Lett., 2011, 13, 3292 CrossRef CAS PubMed.
- (a) Q. Guo, W. Zhu, S. Dong, S. Ma and X. Yan, J. Mol. Struct., 2003, 650, 159 CrossRef CAS; (b) Q. Guo, W. Zhu, S. Ma, D. Yuan, S. Dong and M. Xu, J. Mol. Struct., 2004, 690, 63 CrossRef CAS; (c) Q. Guo, W. Zhu, S. Ma, S. Dong and M. Xu, Polyhedron, 2004, 23, 1461 CrossRef CAS; (d) Q. Guo, W. Zhu, Y. Liu, D. Yuan, J. Zhang and S. Ma, Polyhedron, 2004, 23, 2055 CrossRef CAS; (e) Y. Lu, D.-S. Guo and H.-Y. Zhang, J. Mol. Struct., 2005, 734, 241 CrossRef.
- (a) Y. Bi, W. Liao and H. Zhang, Cryst. Growth Des., 2008, 8, 3630 CrossRef CAS; (b) M. Wu, D. Yuan, Y. Huang, W. Wei, Q. Gao, F. Jiang and M. Hong, Cryst. Growth Des., 2007, 7, 1446 CrossRef CAS; (c) T. Chen, Q. Chen, X. Zhang, D. Wang and L. J. Wan, J. Am. Chem. Soc., 2010, 132, 5598 CrossRef CAS PubMed; (d) N. Kon, N. Iki and S. Miyano, Org. Biomol. Chem., 2003, 1, 751 RSC.
- (a) N. Iki, T. Suzuki, K. Koyama, C. Kabuto and S. Miyano, Org. Lett., 2002, 4, 509 CrossRef CAS PubMed; (b) Y. Liu, D.-S. Guo, E.-C. Yang, H.-Y. Zhang and Y.-L. Zhao, Eur. J. Org. Chem., 2005, 162 CrossRef CAS; (c) Y. Liu, D.-S. Guo and H.-Y. Zhang, J. Mol. Struct., 2005, 734, 241 CrossRef CAS.
- Y. Liu, D.-S. Guo, H.-Y. Zhang, S. Kang and H.-B. Song, Cryst. Growth Des., 2006, 6, 1399 CAS.
- M.-Y. Wu, D.-Q. Yuan, B.-Q. Chen, Y.-G. Huang, F. Jiang and M.-C. Hong, Supramol. Chem., 2007, 19, 411 CrossRef CAS.
- Q.-L. Guo, Y.-F. Deng and X.-Y. Qu, Chinese J. Strut. Chem., 2011, 30, 474 CAS.
- G. William Orr, L. J. Barbour and J. L. Atwood, Science, 1999, 285, 1049 CrossRef.
- K. Uma Maheswara Rao, M. Yamada, T. Kimuro, H. Katagiri, Y. Kondo and F. Hamada, RSC Adv., 2015, 5, 30140 RSC.
- R. C. Johnson and P. H.-Y. Cheong, Org. Biomol. Chem., 2013, 11, 5057 Search PubMed.
- C.-Q. Wan, J. Han and T. C. W. Mak, New J. Chem., 2009, 33, 707–712 RSC.
- (a) N. Iki, T. Fujimoto and S. Miyano, Chem. Lett., 1998, 625 CrossRef CAS; (b) D. Yuan, W. Zhu, S. Ma and X. Yan, J. Mol. Struct., 2002, 616, 241 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary figures and crystallographic data. CCDC 1441358. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra27640c |
| This journal is © The Royal Society of Chemistry 2016 |