
Sorption mechanism(s) of orthophosphate onto Ca(OH)2 pretreated bentonite
Abstract
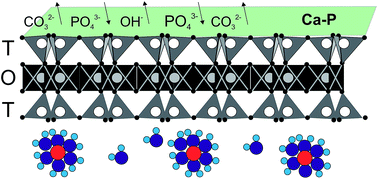
Bentonite was chemically pretreated with Ca(OH)2 to enhance orthophosphate phosphorus (OPP) sorption capacity (qe). The pretreatment resulted in an increase of qe from about 0.3 mg P per g to about 8 mg P per g at pH 7 and 25 °C, and of OPP concentration 100 mg P per L. The effects of solution pH, OPP concentration, sorbent dosage, and temperature on OPP sorption onto pretreated bentonite were investigated. The variation of initial pH (4–9) did not affect qe, however, re-adjustment of the final pH showed that the variation of the final pH had a significant positive effect on qe. The sorption kinetics showed a low rate, reaching half of qe in the first 4 h and equilibrium after 96 h. The calculated Langmuir qmax was 11.68 mg P per g. Thermodynamic analysis suggests that the sorption process is spontaneous and endothermic. Desorption of OPP was high in solutions of 0.1 M HCl and 0.01 EDTA-Na2 and reached 83% and 98%, respectively. We conclude that the predominant sorption mechanism for OPP uptake is inner-sphere surface complexation (ligand exchange). Other less important mechanisms such as surface precipitation and replacement of P5+ for Si4+ on the tetrahedral sites of montmorillonite may operate during the OPP uptake process by pretreated bentonite with Ca(OH)2.
 Please wait while we load your content...
Please wait while we load your content...

