
Highly sensitive electrochemical detection of circulating tumor DNA based on thin-layer MoS2/graphene composites
Abstract
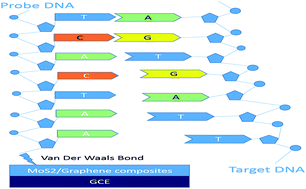
By integrating thin-layer molybdenum disulfide (MoS2) and graphene through a hydrothermal process and an ultrasonic method, a label-free, amplification-free and ultrasensitive circulating tumor DNA electrochemical sensor was made. Compared with other methods, this preparation process was simpler and the electrochemical properties were enhanced. Using a differential pulse voltammetry test, this sensor can detect trace amounts of DNA in the range from 10−16 M to 10−13 M. Compared with other methods that are used to detect the same circulating tumor DNA, this sensor has an obvious advantage in sensitivity, cost and simplicity because it removes the need for labelling processes and amplifiers.
 Please wait while we load your content...
Please wait while we load your content...

