
Synthesis and stability study of isocyano aryl boronate esters and their synthetic applications†
Hao-Ping Fang,
Chia-Chieh Fu,
Chin-Kuen Tai,
Ken-Hao Chang,
Ru-Han Yang,
Meng-Ju Wu,
Hsien-Chi Chen,
Chia-Jung Li,
Shi-Qing Huang,
Wan-Hsiang Lien,
Chih-Hsin Chen,
Chung-Hung Hsieh,
Bo-Cheng Wang,
Siu-Fung Cheung and
Po-Shen Pan *
*
Department of Chemistry, College of Sciences, Tamkang University, No. 151, Yingzhuan Rd., Danshui Dist., New Taipei City 25137, Taiwan. E-mail: popan@mail.tku.edu.tw
First published on 17th March 2016
Abstract
A facile synthesis of isocyano arylboronate esters is reported. Although tri-coordinate boron functional groups are commonly recognized as vulnerable to nucleophilic attack, the newly reported tri-coordinate isocyano arylboronate esters were found to be stable, albeit owing to the presence of an isocyano group. Theoretical calculations, using the DFT/B3LYP/6-31G(d,p) method, revealed that the electron delocalization between the aryl group and the boron atom might contribute to this stability. UV-vis spectroscopic investigations on 2-(4-isocyanophenyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (2a) were in agreement with the theoretical studies, showing a red-shifted absorption compared with that of phenylisonitrile. The reported strategy allows boronate ester substrates to survive throughout two-step operations. Two of the compounds synthesized were successfully exploited in Ugi and Passerini multicomponent reactions to afford corresponding products. In addition, two boron-containing tetrazoles were also prepared under environmentally benign conditions. These results demonstrated that functionalized isocyanides are stable and can be used as strategically as synthetic building blocks.
Introduction
Isocyanides are exceptional moieties that exhibit unique bifunctional reactivity, in which the divalent isocyano carbon can act as a nucleophile or electrophile. Owing to this distinctive feature, isocyanides are frequently used in heterocycle synthesis1 and multicomponent reactions.2 From a synthetic standpoint, the value of isocyanide dramatically increases when an additional functional group is added, because the resulting “functionalized” isocyanide can serve as a multi-purpose synthetic building block, providing opportunities to prepare previously inaccessible targets.3Boronic acid-containing molecules are used in a wide range of organic reactions, including Suzuki–Miyaura coupling,4 Chan–Lam coupling,5 Liebeskind–Srogl coupling,6 conjugate addition,7 homologation,8 electrophilic allyl shifts,9 and C–H coupling reactions.10 Thus, forging boryl and isocyano groups into one molecule could create a highly valuable synthetic material (Fig. 1).
 | ||
| Fig. 1 Potential applications of isocyano boronic acids. | ||
In 1995, van Leusen and co-workers reported the first example of this compound class by converting chloromethylboronates into the corresponding isocyanide (Scheme 1a).11 Although they were able to isolate and characterize the product, they found it hard to handle, requiring storage under nitrogen at −30 °C to avoid rapid decomposition. The observed instability of the boron-substituted methyl isocyanide originated from intermolecular complexation between the electrophilic boron and the nucleophilic isocyano group.12 Previously, our laboratory has successfully employed isocyanide-based Ugi/Passerini multicomponent reactions to construct a series of boron-containing compounds.13 In contrast to van Leusen's approach, we noticed that boron-containing building blocks could be used along with the isocyanide building blocks without jeopardizing the overall reaction outcomes (Scheme 1b–d). These results led us to speculate that the aryl group of the boronic acid substrate could help to reduce the boron's electrophilicity, thus making it less susceptible to complexation with isocyanide. This assumption has prompted us to explore the possibility of synthesizing tri-coordinated boron-substituted phenyl/benzyl isocyanides. Since there are few reports regarding the preparation of these functionalized isocyanides,11,14,15 this proposed approach could serve as a valuable complementary method towards currently limited synthetic strategies.
 | ||
| Scheme 1 (a) Synthesis of boron-substituted methyl isocyanides via van Leusen's strategy; (b) synthesis of boron-containing Ugi-4CR compounds from 3-formylphenyl boronic acids; (c) synthesis of boron-containing Ugi-4CR compounds from (4-(aminomethyl)-2-fluoro)phenyl boronic acid pinacol ester; (d) synthesis of boron-containing Passerini-3CR compounds from 4-carboxyphenyl boronic acid pinacol ester. | ||
Results and discussion
Due to their excellent performance in formylating aniline,16 formic acid (Table 1, entry 1) and sodium ethyl formate/formic acid (Table 1, entry 2) were tested in the formylation of 4-aminophenylboronic acid pinacol ester. Unexpectedly, the results of these reactions were far from ideal, perhaps caused by the pinacol-protected boronic acid of 1a reverting back to the unprotected form under acidic conditions, making it vulnerable to attack from the nucleophilic amino group of the starting material. When ethyl formate was used to formylate 1a under neutral conditions, the yield increased dramatically to 55% (Table 1, entry 3).17 Developed by Cossy and co-workers,18 N-formylsaccharin is a novel formylating reagent capable of formylating amines at ambient temperature under neutral conditions. The most significant feature of N-formylsaccharin is that it chemoselectively formylates amines in the presence of hydroxyl groups. To our delight, we found this reagent suitable for delivering the desired product in moderate yield in just 15 min (Table 1, entry 4, 55%). Increasing the reaction period from to 90 min greatly improved the yield to 95% (Table 1, entries 4–6). Interestingly, changing the solvent from THF to ethyl ether dramatically reduced the yield to 31% (Table 1, entry 7). This observation indicated that the reaction was highly sensitive to solvent polarity. Similarly to THF, acetone afforded an excellent yield after 90 min (Table 1, entry 8, 93%). The yield was not significantly affected when the reaction time was reduced to 60 min (Table 1, entry 9, 96%); however, when the reaction time was further decreased to 30 min, a notable decrease in yield was observed (Table 1, entry 10, 80%). We were pleased to find that, in dichloromethane, the reaction period could be dramatically reduced from 90 to 10 min while maintaining an excellent yield (Table 1, entries 11–13, 98%).
|
|||||
|---|---|---|---|---|---|
| Entry | Formylating agent | Solvent | Temp | Time [min] | Yield [%] |
| a 1a was previously synthesized via a different synthetic strategy.19 | |||||
| 1 | Formic acid | EtOAc | r.t. | 20 | <1 |
| 2 | HCOONa/formic acid | Free | r.t. | 270 | 21 |
| 3 | Ethyl formate | Free | 60 °C | 360 | 55 |
| 4 | N-Formylsaccharin | THF | r.t. | 15 | 55 |
| 5 | N-Formylsaccharin | THF | r.t. | 60 | 73 |
| 6 | N-Formylsaccharin | THF | r.t. | 90 | 95 |
| 7 | N-Formylsaccharin | Et2O | r.t. | 90 | 31 |
| 8 | N-Formylsaccharin | Acetone | r.t. | 90 | 93 |
| 9 | N-Formylsaccharin | Acetone | r.t. | 60 | 95 |
| 10 | N-Formylsaccharin | Acetone | r.t. | 30 | 80 |
| 11 | N-Formylsaccharin | CH2Cl2 | r.t. | 90 | 90 |
| 12 | N-Formylsaccharin | CH2Cl2 | r.t. | 30 | 99 |
| 13 | N-Formylsaccharin | CH2Cl2 | r.t. | 10 | 98 |

With the optimized N-formylation conditions in hand (Table 1, entry 13), seven additional N-formylated boron-substituted benzyl amines were synthesized (1b–h), with the results are summarized in Table 2. The yields ranged from 45–79%.
|
|||
|---|---|---|---|
| Entry | R | Product | Yield [%] |
| a All reactions were carried out using boron-substituted aryl amine (1.0 equiv.), N-formylsaccharin (1.1 equiv.) in dichloromethane (0.2 M). | |||
| 1 |  |
1b | 74 |
| 2 |  |
1c | 78 |
| 3 |  |
1d | 57 |
| 4 |  |
1e | 67 |
| 5 |  |
1f | 45 |
| 6 |  |
1g | 43 |
| 7 | 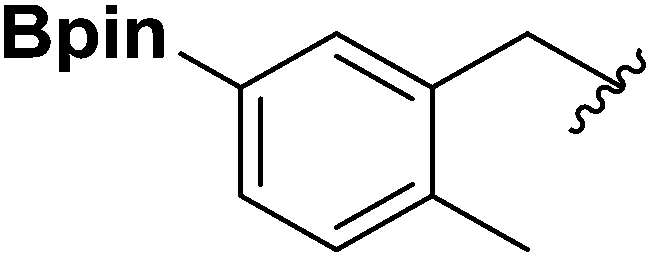 |
1h | 54 |

Three representative dehydration protocols15,20 were evaluated; the results are summarized in Table 3. Compared with other traditional dehydrating reagents, propylphosphonic anhydride (®T3P) has several significant advantages, including low toxicity and generally delivering high product yields. Furthermore, products generated by ®T3P can be purified via simple extraction due to the ®T3P byproduct being water soluble.21 Thus, it was chosen an ideal dehydrating agent to convey the boron-substituted isocyanides. Unfortunately, the use of ®T3P resulted in low yields of the desired products (Table 3, entry 1, 18%). Porcheddu et al. reported a facile microwave-assisted synthesis using cyanuric chloride as the dehydrating agent,22 which went to completion in under 10 min at 50 °C. Under these conditions, we were able to obtain the desired product in much higher yield (Table 3, entry 2, 66%). This improved yield at a lower temperature caused us to speculate that formation of the desired product was highly dependent on reaction temperature. It is well documented that the dehydration of N-formamide using POCl3 takes place at low temperatures, thus representing a promising option for the synthesis of the desired isocyanides. Indeed, we were able to obtain desired product 2a in excellent yield (98%, Table 3, entry 3).
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Dehyd. agent | Base | Solvent | Temp. [°C] | Conc. [M] | Time [min] | Yield [%] |
| 1 | ®T3P | TEA | CH2Cl2 | 80 | 0.5 | 300 | 18 |
| 2 | TCT | TEA | CH2Cl2 | 50 | 0.5 | 3 | 66 |
| 3 | POCl3 | TEA | CH2Cl2 | 0 | 0.5 | 60 | 98 |

The structure of 2a was determined unambiguously by X-ray crystallography (Fig. 2). To the best of our knowledge, this is the first report of the crystal structure of a boron-substituted phenyl isocyanide.
 | ||
| Fig. 2 X-ray crystal structures of 2a, thermal ellipsoids drawn at the 50% probability level. | ||
By utilizing the optimized conditions (Table 3, entry 3), seven additional boron-substituted isocyanides were synthesized accordingly (2b–h). Results are shown in Table 4.
|
|||
|---|---|---|---|
| Entry | R | Product | Yield [%] |
| 1 | 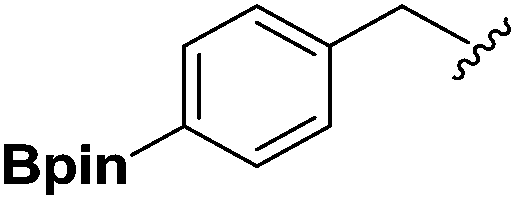 |
2b | 80 |
| 2 |  |
2c | 88 |
| 3 | 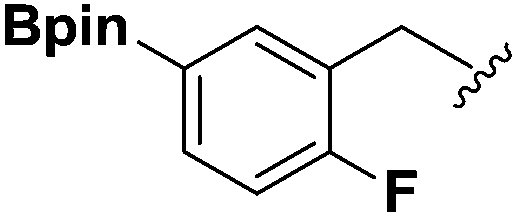 |
2d | 58 |
| 4 |  |
2e | 65 |
| 5 | 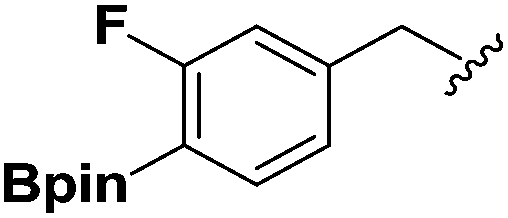 |
2f | 52 |
| 6 |  |
2g | 75 |
| 7 |  |
2h | 71 |

Unlike highly unstable boron-substituted methyl isocyanides, the boron-substituted phenyl and benzyl isocyanides produced in this study (Table 4) could be stored at ambient temperature without a N2 atmosphere for one week (for a month if stored at 0 °C). In order to investigate how phenyl groups contributed to this improved stability, a series of theoretical calculations were performed using a DFT/B3LYP/6-31G(d,p) method by controlling the dihedral angle between the phenyl group and boron functional group. It was found that when the dihedral angle between the phenyl and boron functional groups in 2a was 90.00°, the electron density of the phenyl group did not delocalize to the boron functional group (Table 5, entry 1, LUMO state). However, when the dihedral angle changed to 15.95° (based on X-ray crystallography of 2a), delocalization of the electron density from the phenyl group to the boron functional group was observed (Table 5, entry 2, LUMO state). This result was also observed when 2a was at the optimized dihedral angle of 2.74° (Table 5, entry 3).
| Entry | Dihedral angle | HOMO | LUMO |
|---|---|---|---|
| 1 | 90.00° | 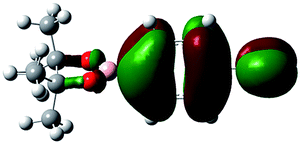 |
 |
| 2 | 15.95° |  |
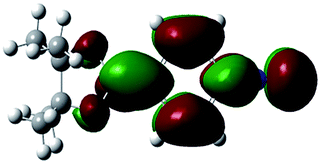 |
| 3 | 2.74° |  |
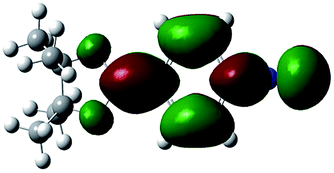 |
Projected density of state (pDOS) analysis of the HOMO and LUMO states of 2a was also retrieved using the DFT/B3LYP/6-31G(d,p) method; the results are shown in Table 6. It was found that when the dihedral angle between the phenyl group and the boron functional group was 90.00°, and the relative charge density of boron was 1.3 in the LUMO state (Table 6, entry 1), while at 15.95°, the value increased noticeably to 6.7 (Table 6, entry 2). This indicated that an electron delocalization event would occur when p-orbitals of the aryl group and boron atom aligned properly. Similar results were also obtained with the optimal dihedral angle, where the value was 6.8 (Table 6, entry 3).
| Entry | Dihedral angle | HOMO | LUMO | ||||||
|---|---|---|---|---|---|---|---|---|---|
| OO | B | ph | NC | OO | B | ph | NC | ||
| 1 | 90.00° | 4.2 | 0.8 | 76.7 | 18.3 | 5.5 | 1.3 | 72.9 | 20.2 |
| 2 | 15.95° | 8.4 | 0.3 | 73.8 | 17.5 | 8.1 | 6.7 | 69.2 | 16.0 |
| 3 | 2.74° | 8.8 | 0.3 | 73.5 | 17.4 | 7.9 | 6.8 | 69.4 | 15.9 |
The UV-vis spectroscopic study was employed to validate the abovementioned theoretical prediction (Fig. 3). The maximum absorption of phenylisonitrile at 228 nm was assigned to the HOMO–LUMO transition based on theoretical predictions. It was found that after attaching a boron functional group at the para-position (2a), this absorption band red-shifted to 237 nm. Such a phenomenon could be recognized as evidence of the establishment of a conjugation system in 2a. This observation was also in agreement with theoretical calculations.
 | ||
| Fig. 3 UV-vis spectrum of phenylisonitrile and 2a. | ||
Therefore, the improved stability of isocyano arylboronate esters could be attributed to the delocalization of electrons from the phenyl group to the electrophilic boron atom. Hence, this extended conjugation system made the tri-coordinate boron functional group more resistant to nucleophilic attack from the isocyano group. This conjugation effect was also observed by Mayr and co-workers, who found that electrophilic aldehydes attached to conjugated systems were less prone to nucleophilic attack compared with those without conjugated systems.23
Due to the successes of Bortezomib (Fig. 4a)24 and Kerydin (Fig. 4b),25 the development of boron-containing pharmaceutical agents has progressed significantly (Fig. 4c–f).26 The current strategies for making these agents mainly depend on linear27 or convergent approaches.28 Despite their different synthetic tactics, they all have a limitation in common, where the position of the boron group must be pre-determined. Any attempt to re-position the boron group was synthetically demanding. Given that the biological activities of these compounds was highly dependent on the location of the boron group, for applications in drug discovery programs, it would be ideal to find a way of generating analogues that have a common core structure, but with the boron group revolving around it.
 | ||
| Fig. 4 Boron-containing pharmaceuticals. | ||
Previously, our laboratory had reported the use of isocyanide-based multicomponent reactions to accomplish this objective.13 By simply exchanging the boron-substituted building blocks, we were able to relocate the boron group within the molecular framework without revamping the synthetic strategy. This strategy; however, was not fully realized due to the exclusion of isocyano arylboronate esters. By introducing previously inaccessible boron-substituted isocyanides, this strategy was now validated. The results are shown in Scheme 2a and b.
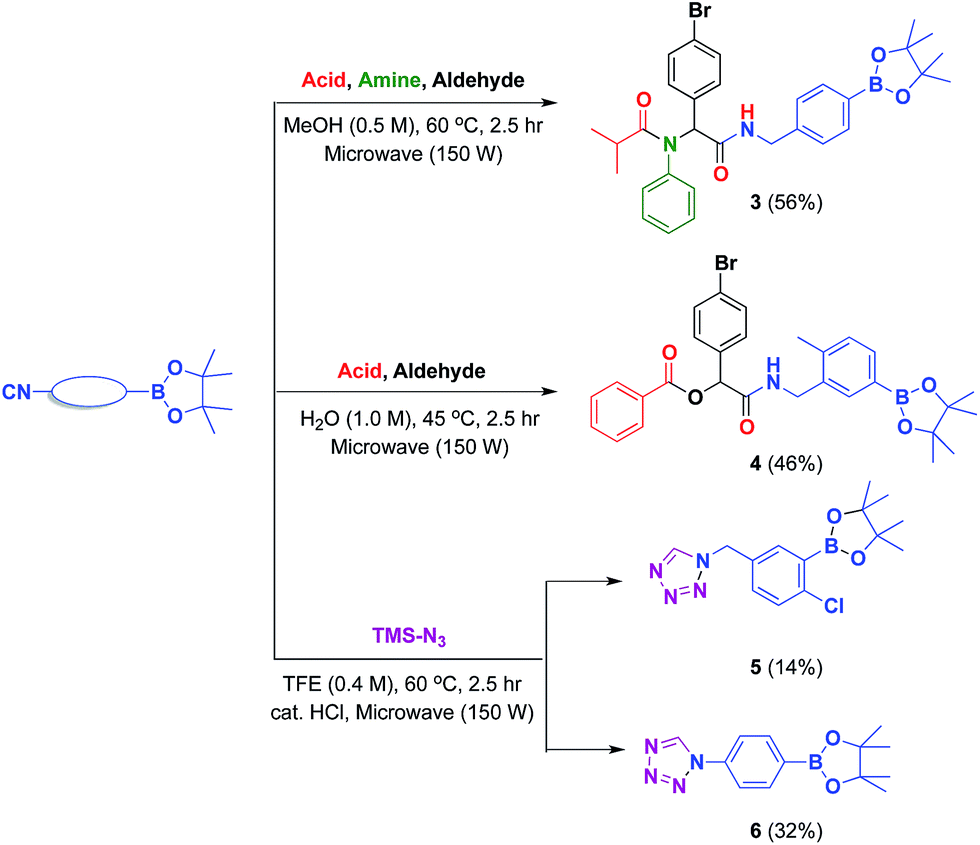 | ||
| Scheme 2 Applications of isocyano aryl boronate esters. | ||
The reported isocyanides could not only be utilized in multicomponent reactions, but also in heterocycle synthesis. Among heterocycles, tetrazole is of particular interest.29 Compared with peptides substrates, tetrazoles tend to have an improved absorption profile with greater metabolic stability. In this report, we have developed microwave-assisted conditions that allowed the preparation of two boron-containing tetrazoles 5 and 6 (Scheme 2c). These tetrazoles were stable and easily obtained in crystalline form in CH2Cl2/n-hexane solution. The structures of 5 and 6 (ref. 30) were later confirmed by single crystal X-ray diffraction (Fig. 5 and 6).
 | ||
| Fig. 5 X-ray crystal structure of 5, thermal ellipsoids drawn at the 50% probability level. | ||
 | ||
| Fig. 6 X-ray crystal structure of 6, thermal ellipsoids drawn at the 50% probability level. | ||
Experimental section
General methods and materials
All starting materials were obtained from commercial suppliers and used without further purification unless otherwise noted. Reactions were performed in a CEM Discover Benchmate™ microwave reactor with sealed vessels. Temperatures of the microwave-assisted reactions were monitored using an external IR sensor. Unless otherwise specified 1H and 13C NMR spectra were recorded on a Bruker AC-300 FT-NMR spectrometer at 300 and 75.5 MHz, respectively. 19F NMR spectra were recorded on a Bruker AVIII 400 FT-NMR spectrometer at 376.5 MHz. 11B NMR spectra were recorded on a Bruker Avance 600 FT-NMR spectrometer at 192.5 MHz. All 11B chemical shifts were referenced to external BF3·OEt2 (0.0 ppm). Data are represented as follows: chemical shifts (ppm), multiplicity (s: singlet, d: doublet, t: triplet, m: multiplet, br: broad), coupling constant J (Hz). Melting points were determined using a Fargo MP-2D melting point apparatus and are uncorrected. High-resolution ESI mass spectra were obtained using a Finnigan MAT 95S instrument. X-ray crystallography data was obtained using a Bruker APEX II X-ray Single Crystal Diffractometer. UV-vis spectroscopy data was collected using a Thermo Scientific Evolution 60S UV-Visible Spectrophotometer.General procedure for the preparation of boron-substituted formamides

![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :8) eluent, to afford the desired product as a brown solid in 98% yield (242.00 mg); mp 111 °C; 1H-NMR (600 MHz, CDCl3) δ 8.78 (d, J = 11.4 Hz, 0.5H), 8.45–8.40 (br, 1H), 8.37 (s, 0.5H), 7.77 (t, 2H), 7.54 (d, J = 8.4 Hz, 1H), 7.08 (d, J = 8.4 Hz, 1H), 1.39 (s, 12H); 13C-NMR (150 MHz, CDCl3) δ 162.2, 158.9, 139.4, 139.3, 136.4, 135.8, 118.7, 117.1, 83.9, 83.7, 24.82; 11B-NMR (192.5 MHz, CDCl3) δ 30.81. HRMS (ESI): m/z calcd for C13H19O3NB: 248.1453 [M + H]+; found: 248.1449.
:8) eluent, to afford the desired product as a brown solid in 98% yield (242.00 mg); mp 111 °C; 1H-NMR (600 MHz, CDCl3) δ 8.78 (d, J = 11.4 Hz, 0.5H), 8.45–8.40 (br, 1H), 8.37 (s, 0.5H), 7.77 (t, 2H), 7.54 (d, J = 8.4 Hz, 1H), 7.08 (d, J = 8.4 Hz, 1H), 1.39 (s, 12H); 13C-NMR (150 MHz, CDCl3) δ 162.2, 158.9, 139.4, 139.3, 136.4, 135.8, 118.7, 117.1, 83.9, 83.7, 24.82; 11B-NMR (192.5 MHz, CDCl3) δ 30.81. HRMS (ESI): m/z calcd for C13H19O3NB: 248.1453 [M + H]+; found: 248.1449.




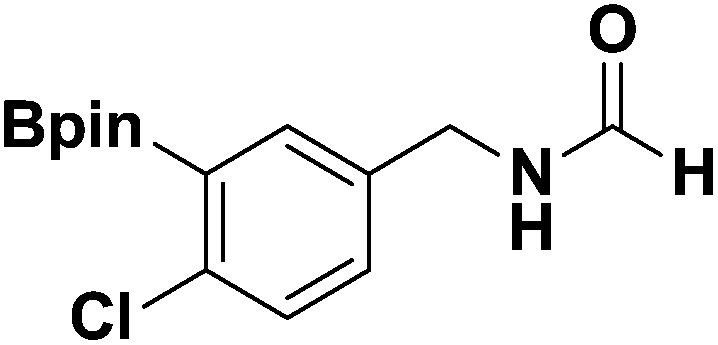

General procedure for the preparation of boron-substituted isocyanides




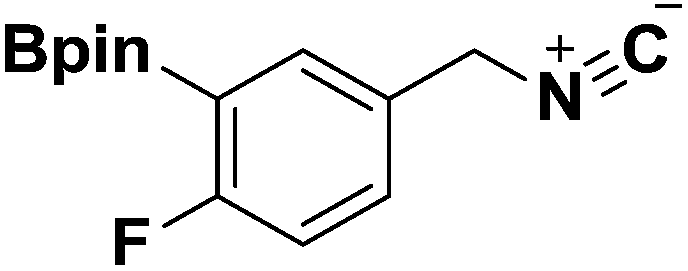

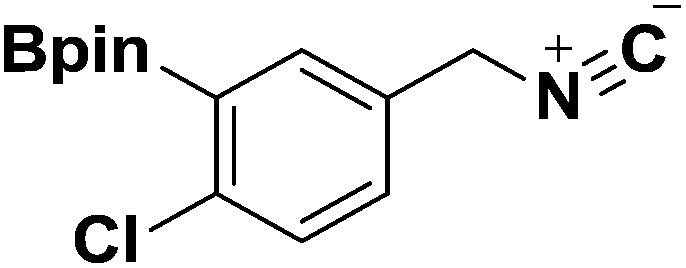

General procedure for the preparation of Ugi-4CR analogues
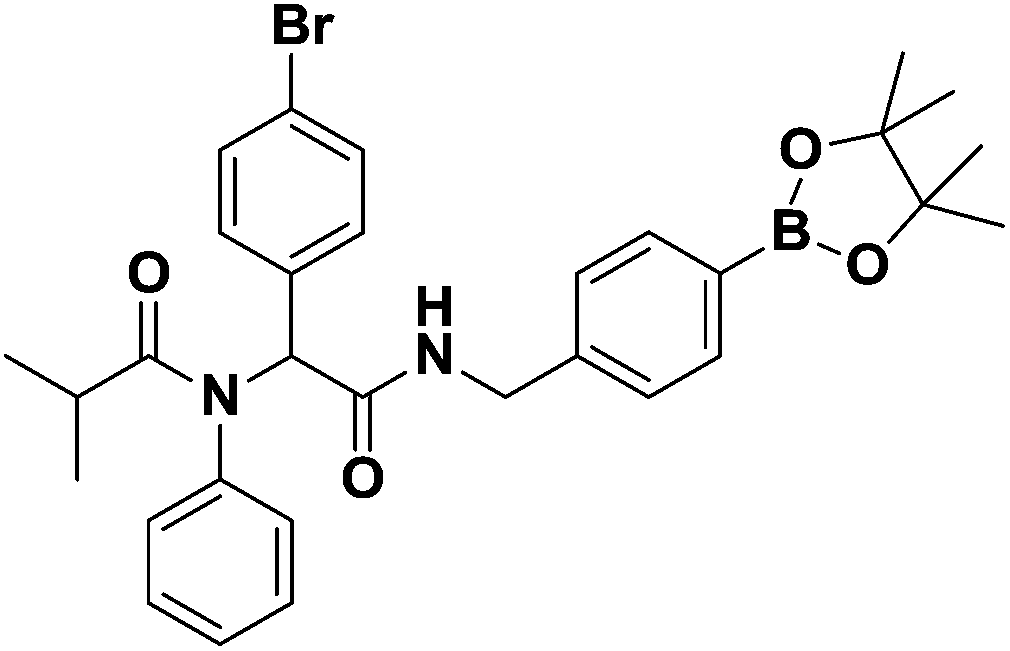 :7) eluent to afford the desired product as a colorless oil in 56% yield (301.00 mg); 1H-NMR (Bruker AC-600 FT-NMR spectrometer at 600 MHz, CDCl3) δ 7.78 (d, J = 7.8 Hz, 1H), 7.73 (d, J = 7.8 Hz, 2H), 7.31–7.20 (d, 8H), 6.99 (d, J = 8.4 Hz, 2H), 6.49 (br, NH), 6.01 (s, 1H), 4.49 (s, 2H), 2.38 (m, 1H), 1.33 (s, 12H), 1.01 (q, J = 6.6, 6H); 13C-NMR (Bruker AC-600 FT-NMR spectrometer at 150.9 MHz, CDCl3) δ 178.1, 169.3, 141.1, 139.7, 135.2, 135.0, 134.4, 133.4, 131.9, 130.2, 130.1, 129.0, 128.3, 128.2, 127.0, 126.7, 122.6, 83.7, 64.2, 43.6, 31.8, 31.8, 24.8, 19.6; 11B-NMR (CDCl3) δ 30.79; HRMS (ESI): m/z calcd for C31H37BBrN2O4: 591.2024 [M + H]+; found: 591.2024.
:7) eluent to afford the desired product as a colorless oil in 56% yield (301.00 mg); 1H-NMR (Bruker AC-600 FT-NMR spectrometer at 600 MHz, CDCl3) δ 7.78 (d, J = 7.8 Hz, 1H), 7.73 (d, J = 7.8 Hz, 2H), 7.31–7.20 (d, 8H), 6.99 (d, J = 8.4 Hz, 2H), 6.49 (br, NH), 6.01 (s, 1H), 4.49 (s, 2H), 2.38 (m, 1H), 1.33 (s, 12H), 1.01 (q, J = 6.6, 6H); 13C-NMR (Bruker AC-600 FT-NMR spectrometer at 150.9 MHz, CDCl3) δ 178.1, 169.3, 141.1, 139.7, 135.2, 135.0, 134.4, 133.4, 131.9, 130.2, 130.1, 129.0, 128.3, 128.2, 127.0, 126.7, 122.6, 83.7, 64.2, 43.6, 31.8, 31.8, 24.8, 19.6; 11B-NMR (CDCl3) δ 30.79; HRMS (ESI): m/z calcd for C31H37BBrN2O4: 591.2024 [M + H]+; found: 591.2024.General procedure for the preparation of Passerini-3CR analogues
 :7) to afford the desired product as a yellow colored solid in 46% yield (84.40 mg), mp: 162 °C; 1H-NMR (Bruker AC-600 FT-NMR spectrometer at 600 MHz, CDCl3) δ 8.04 (d, J = 7.8 Hz, 2H), 7.65–7.42 (m, 9H), 7.19 (d, J = 7.2 Hz, 1H), 6.29 (s, 1H), 6.27 (br, NH), 4.58–4.44 (dd, J = 5.4, 46.8 Hz, 2H), 2.25 (s, 3H), 1.33 (s, 12H); 13C-NMR (Bruker AC-600 FT-NMR spectrometer at 150.9 MHz, CD3OD) δ 167.4, 164.8, 140.2, 135.0, 134.6, 134.4, 133.7, 131.9, 130.2, 129.8, 128.9, 128.6, 123.1, 83.8, 75.2, 41.8, 24.8, 19.1; 11B-NMR (CDCl3) δ 30.87. HRMS (ESI): m/z calcd for C29H32BBrNO5: 564.1551 [M + H]+; found: 564.1552.
:7) to afford the desired product as a yellow colored solid in 46% yield (84.40 mg), mp: 162 °C; 1H-NMR (Bruker AC-600 FT-NMR spectrometer at 600 MHz, CDCl3) δ 8.04 (d, J = 7.8 Hz, 2H), 7.65–7.42 (m, 9H), 7.19 (d, J = 7.2 Hz, 1H), 6.29 (s, 1H), 6.27 (br, NH), 4.58–4.44 (dd, J = 5.4, 46.8 Hz, 2H), 2.25 (s, 3H), 1.33 (s, 12H); 13C-NMR (Bruker AC-600 FT-NMR spectrometer at 150.9 MHz, CD3OD) δ 167.4, 164.8, 140.2, 135.0, 134.6, 134.4, 133.7, 131.9, 130.2, 129.8, 128.9, 128.6, 123.1, 83.8, 75.2, 41.8, 24.8, 19.1; 11B-NMR (CDCl3) δ 30.87. HRMS (ESI): m/z calcd for C29H32BBrNO5: 564.1551 [M + H]+; found: 564.1552.General procedure for the preparation of boron-containing tetrazoles

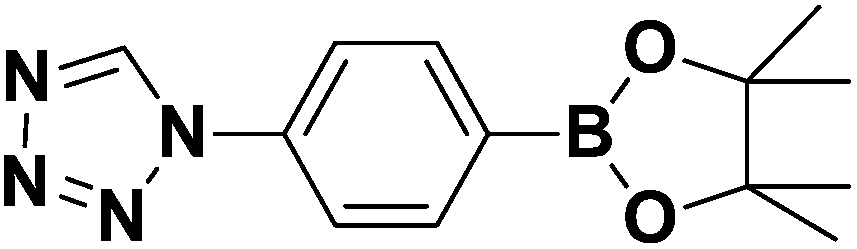
Conclusions
In conclusion, we successfully developed a synthetic strategy for the preparation of boron-substituted phenyl/benzyl isocyanides. A combination of X-ray crystallography data, theoretical calculations, and UV-spectroscopic studies provided direct evidence of electron delocalization in these compounds. Delocalization sufficiently reduced the electrophilicity of boron, making them stable enough for storage at ambient temperature for one week. The application of the reported products to isocyanide-based Ugi-4CR/Passerini-3CR, as well as tetrazole formation, was also described. These results demonstrate the stability of the reported boron-substituted phenyl/benzyl isocyanides, and their applications in various types of synthetic reaction.Acknowledgements
This research was supported by the Ministry of Science and Technology in Taiwan (MOST-103-2113-M-032-010, MOST-104-2113-M-032-001). We thank Professor Hsiu-Fu Hsu from Department of Chemistry of Tamkang University for insightful discussions, the Department of Chemistry of Tamkang University for equipment and financial support, Ting-Shen Kuo from the Department of Chemistry of National Taiwan Normal University for conducting X-ray experiments, Professor Chiung-Cheng Huang from Department of Chemical Engineering of Tatung University and Dr. Yi-Jhen Feng from the Department of Chemistry of National Taiwan University for conducting 19F NMR experiments, and Shen-Shen Chen from the Department of Chemistry of Tamkang University for conducting 11B-NMR experiments.Notes and references
- (a) K. Kobayashi, R. Nakahashi, A. Takanohashi, T. Kitamura, O. Morikawa and H. Konishi, Chem. Lett., 2002, 30, 624–625 CrossRef; (b) Y. Tsunenishi, H. Ishida, K. Itoh and M. Ohno, Synlett, 2000, 9, 1318–1320 Search PubMed.
- (a) A. Dömling and I. Ugi, Angew. Chem., Int. Ed., 2000, 39, 3168–3210 CrossRef; (b) A. Dömling, Chem. Rev., 2006, 106, 17–89 CrossRef PubMed; (c) I. Ugi, A. Domling and B. J. Werner, Heterocycl. Chem., 2000, 37, 647–658 CrossRef CAS.
- (a) D. van Leusen and A. M. van Leusen, Org. React., 2003, 57, 419 Search PubMed; (b) A. M. van Leusen, J. Wildeman and O. H. Oldenziel, J. Org. Chem., 1977, 42, 1153–1159 CrossRef CAS; (c) A. M. van Leusen, Heterocycl. Chem., 1980, 5, S-111 CAS; (d) J. S. Andrew, J. Kassick, M. Mellinger, J. J. Filan, A. Allen and M. A. Olsen, J. Org. Chem., 2000, 65, 1516–1524 CrossRef.
- B. H. Lipshutz, T. B. Petersen and A. R. Abela, Org. Lett., 2008, 10, 1333–1336 CrossRef CAS PubMed; J. Han, Y. Liu and R. Guo, J. Am. Chem. Soc., 2009, 131, 2060–2061 CrossRef PubMed; C. Baillie, L. Zhang and J. Xiao, J. Org. Chem., 2004, 69, 7779–7782 CrossRef PubMed; B. Saito and G. C. Fu, J. Am. Chem. Soc., 2007, 129, 9602–9603 CrossRef PubMed; A. F. Littke, C. Dai and G. C. Fu, J. Am. Chem. Soc., 2000, 122, 4020–4028 CrossRef; D. D. Dolliver, B. T. Bhattarai, A. Pandey, M. L. Lanier, A. S. Bordelon, S. Adhikarl, J. A. Dinser, P. F. Flowers, V. S. Wills, C. L. Schneider, K. H. Shaughnessy, J. N. Moore, S. M. Raders, T. S. Snowden, A. S. McKim and F. R. Fronczek, J. Org. Chem., 2013, 78, 3676–3687 CrossRef PubMed.
- (a) D. M. T. Chan, K. L. Monaco, R. Li, D. Bonne, C. G. Clark and P. Y. S. Lam, Tetrahedron Lett., 2003, 44, 3863–3865 CrossRef CAS; (b) P. Y. S. Lam, G. Vincent, D. Bonne and C. G. Clark, Tetrahedron Lett., 2003, 44, 4927–4931 CrossRef CAS.
- (a) L. S. Liebeskind and J. Srogl, J. Am. Chem. Soc., 2000, 122, 11260–11261 CrossRef CAS; (b) Y. Yu and L. S. Liebeskind, J. Org. Chem., 2004, 69, 3554–3557 CrossRef CAS PubMed; (c) J. M. Villalobos, J. Srogl and L. S. Liebeskind, J. Am. Chem. Soc., 2007, 129, 15734–15735 CrossRef CAS PubMed.
- J. D. Sieber, S. Liu and J. P. Morken, J. Am. Chem. Soc., 2007, 129, 2214–2215 CrossRef CAS PubMed.
- D. S. Matteson, K. M. Sadhu and M. L. Peterson, J. Am. Chem. Soc., 1986, 108, 810–819 CrossRef CAS.
- F. Peng and D. G. Hall, J. Am. Chem. Soc., 2007, 129, 3070–3071 CrossRef CAS PubMed.
- (a) J. Takagi, K. Sato, J. F. Hartwig, T. Ishiyama and N. Miyaura, Tetrahedron Lett., 2002, 43, 5649–5651 CrossRef CAS; (b) T. Ishiyama, J. Takagi, K. Ishida, N. Miyaura, M. R. Anastasi and J. F. Hartwig, J. Am. Chem. Soc., 2002, 124, 390–391 CrossRef CAS PubMed; (c) T. Ishiyama, Y. Nobuta, J. F. Hartwig and N. Miyaura, Chem. Commun., 2003, 2924–2925 RSC.
- J. P. G. Versleijen, P. M. Faber, H. H. Bodewes, A. H. Braker, D. van Leusen and A. M. van Leusen, Tetrahedron Lett., 1995, 36, 2109–2112 CrossRef CAS.
- H. Witte, P. Mischke and G. Hesse, Leibigs Ann. Chem., 1969, 722, 21–28 CrossRef CAS.
- R. C. Lian, M. H. Lin, P. H. Liao, J. J. Fu, M. J. Wu, Y. C. Wu, F. R. Chang, C. C. Wu and P. S. Pan, Tetrahedron, 2014, 70, 1800–1804 CrossRef CAS; C. C. Chai, P. Y. Liu, C. H. Lin, H. C. Chen, Y. C. Wu, F. R. Chang and P. S. Pan, Molecules, 2013, 18, 9488–9511 CrossRef PubMed; S. H. Chung, T. J. Lin, Q. Y. Hu, C. H. Tsai and P. S. Pan, Molecules, 2013, 18, 12346–12367 CrossRef PubMed.
- E. S. Priestley and C. P. Decicco, Org. Lett., 2000, 2, 3095–3097 CrossRef CAS PubMed.
- A. Zajdlik, Z. Wang, J. L. Hickey, A. Aman, A. D. Schimmer and A. K. Yudin, Angew. Chem., Int. Ed, 2013, 52, 8411–8415 CrossRef CAS PubMed.
- H. S. Mosa and H. Sharghi, J. Org. Chem., 2006, 71, 6652–6654 CrossRef PubMed.
- K. P. Dhake, P. J. Tambade, R. S. Singhal and B. M. Bhanage, Green Chem. Lett. Rev., 2011, 4, p151–157 CrossRef.
- (a) T. Cochet, V. Bellosta, A. Greiner, D. Roche and J. Cossy, Synlett, 2011, 13, 1920–1922 Search PubMed; (b) T. Ueda, H. Konishi and K. Manabe, Angew. Chem., Int. Ed., 2013, 52, 8611–8615 CrossRef CAS PubMed.
- A. M. Chaudhari, D. Dhanak, S. D. Knight, D. J. Morgans Jr and C. A. Parrish, P. C. T. Patent, WO2006020358 A2, 2006.
- E. S. Priestley and C. P. Decicco, Org. Lett., 2000, 2, 3095–3097 CrossRef CAS PubMed.
- (a) J. Klose, M. Bienert, C. Mollenkopf, D. Wehle, C. W. Zhang, L. A. Carpino and P. Henklein, Chem. Commun., 1999, 1847–1848 RSC; (b) F. L. Zumpe, M. Flüβ, K. Schmitz and A. Lender, Tetrahedron Lett., 2007, 48, 1421–1423 CrossRef CAS.
- A. Porcheddu, G. Giacomelli and M. Salaris, J. Org. Chem., 2005, 70, 2361–2363 CrossRef CAS PubMed.
- R. Appel and H. Mayr, J. Am. Chem. Soc., 2011, 133, 8240–8251 CrossRef CAS PubMed.
- J. Adams and M. Kauffman, Cancer Invest., 2004, 22, 304–311 CrossRef CAS PubMed.
- S. J. Baker, Y. K. Zhang, T. Akama, A. Lau, H. Zhou, V. Hernandez, W. Mao, M. R. K. Alley, V. Sanders and J. J. Plattner, J. Med. Chem., 2006, 49, 4447–4450 CrossRef CAS PubMed.
- S. J. Baker, C. Z. Ding, T. Akama, Y. K. Zhang, V. Hernandez and Y. Xia, Future Med. Chem., 2009, 1, 1275–1288 CrossRef CAS PubMed.
- (a) Y. Zhu, S. Yao, B. Xu, Z. Ge, J. Cui, T. Cheng and R. Li, Bioorg. Med. Chem., 2009, 17, 6851–6861 CrossRef CAS PubMed; (b) M. A. Beenen, C. An and J. A. Ellman, J. Am. Chem. Soc., 2008, 130, 6910–6911 CrossRef CAS PubMed; (c) J. A. Adams, M. Behnke, A. A. Cruickshank, L. R. Dick, L. Grenier, J. M. Klunder, Y. T. Ma, L. Plamondon and R. L. Stein, Bioorg. Med. Chem. Lett., 1998, 8, 333–338 CrossRef CAS PubMed.
- A. S. Ivanov, A. A. Zhalnina and S. V. Shishkov, Tetrahedron, 2009, 65, 7105–7108 CrossRef CAS.
- A. Malek and A. Sarvary, RSC Adv., 2015, 5, 60938–60955 RSC.
- D. Xiao, Y. Zhu, Y. Hu, H. Wang, Y. Liu, J. Li, D. Sun, Z. Wang, Y. Wei, Z. Wang, G. Tang and L. Jing, P. C. T. Patent, WO2012103806 A1, 2012.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1443748–1443750. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra27624a |
| This journal is © The Royal Society of Chemistry 2016 |