
An efficient, scalable synthesis of ferrocenylphosphine and dichloroferrocenylphosphine†
Abstract
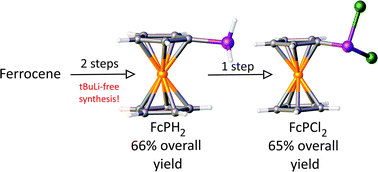
A new synthetic route to FcPH2 and FcPCl2 (Fc = ferrocenyl) is presented. This method avoids the challenging monolithiation of ferrocene, as well as any tedious purification steps. All reactions are high yielding and easily conducted on a relatively large scale, using economical and commercially available synthetic precursors.
 Please wait while we load your content...
Please wait while we load your content...

