DOI:
10.1039/C5RA27583K
(Paper)
RSC Adv., 2016,
6, 33177-33183
Effects of sulfur compounds on the hydrogenation and isomerization of 1-hexene over a sulfided CoMo catalyst for hydrodesulfurization†
Received
23rd December 2015
, Accepted 17th March 2016
First published on 30th March 2016
Abstract
The transformation of olefins is crucial to the hydrodesulfurization (HDS) of fluid catalytic cracking (FCC) gasoline. The reactivity of 1-hexene was studied over a sulfided CoMo/Al2O3 catalyst. Effects of hydrogen sulfide (H2S) and three organic sulfur compounds on 1-hexene transformation were also investigated. As for the hydroconversion of 1-hexene, the double-bond isomerization (ISOM) of 1-hexene proceeded readily at lower temperatures while the hydrogenation (HYD) of 1-hexene and its isomers predominated at higher temperatures. The inhibiting effect of H2S on both isomerization and hydrogenation reactions resulted in the decline in 1-hexene HYD conversion. The saturation rate of 1-hexene increased sharply when H2S content decreased to below 100 μg g−1 S. It was also found that thiophene (T), 2-methylthiophene (2MT) and 3-methylthiophene (3MT) strongly inhibited the hydrogenation of 1-hexene. Furthermore, the inhibiting effect on 1-hexene HYD reaction originated from H2S generated by organic sulfide desulfurization. More isomerization products were obtained in the presence of sulfur compounds, probably attributable to the inhibition of sulfur compounds on the hydrogenation of 1-hexene and its isomers.
Introduction
Recent stringent environmental regulations require the production of ultralow sulfur gasoline fuel (S < 10 ppm). Fluid catalytic cracking (FCC) gasoline, which contains about 20–40 vol% olefins as well as high levels of sulfur,1 is the major blending component in the gasoline pool. Hydrotreating is one of the most widely used refining process to remove sulfur from FCC gasoline. Organic sulfur compounds in FCC gasoline are relatively reactive under conventional hydrodesulfurization (HDS) conditions. The principle problem with HDS of FCC gasoline is that octane number decreases significantly during the hydrotreating process due to the saturation of olefins.2 Consequently, the best way of achieving the ultra-deep HDS with minimizing octane loss is to suppress the reactivity of olefins in the HDS process.
Numerous efforts have been devoted to HDS, such as developing highly active HDS catalysts3–9 and studying on the active phase structure.10–16 However, investigations concerning the reactivity of olefins are scarce.17–20 During the HDS process of FCC gasoline, hydrogen sulfide (H2S) can be created in the reaction system. Moreover, H2S contributes to maintain the sulfidation state of the catalysts. However, as for the effect of H2S on the transformation of olefins over sulfided Mo-based catalysts, contradictory results exist in the literature. Xi et al.21 found that H2S in circulating hydrogen promoted the hydrogenation saturation of olefins. Lamic et al.22 claimed that olefin hydrogenation activity of the NiMo catalyst was inhibited by the presence of H2S, whereas those of CoMo and MoS2 catalysts were weakly or not affected. It was generally admitted that the steric effects of both olefin structure and MoS2 crystalline structure, and the state of Mo coordinatively unsaturated sites (CUS) or so-called sulfur vacancies on sulfide phases were closely related to the hydroconversion of olefins.18,23 In our previous work,7 we found that the alumina with better crystallinity and less hydroxyl groups stimulated the promoting effect of cobalt on molybdenum and contributed to a noticeable increase in HDS activity. In this study, we prepared the CoMo/Al2O3 catalyst with this kind of alumina and investigated the transformation of 1-hexene over the sulfided CoMo/Al2O3 catalyst in the presence of H2. Effects of H2S and organic sulfur compounds on 1-hexene hydroconversion were also systematically examined. All experiments were carried out under usual HDS operating conditions.
Experimental
A commercial alumina with better crystallinity and less hydroxyl groups was used as the support. The CoMo/Al2O3 catalyst was prepared by the incipient wetness impregnation method with the aqueous solution containing the Co, Mo salts and chelating agent, followed by drying at 393 K for 2 h. The final metal loadings on the catalyst were 12.2 wt% of MoO3 and 4.9 wt% of CoO.
The surface areas and pore structures of samples were determined by N2 physisorption. Surface hydroxyl groups of alumina were determined by pyridine adsorption Fourier-transform infrared (Py-IR). The metal loadings of the catalyst were measured by X-ray fluorescence (XRF). The sulfidation degree and MoS2 morphology of the sulfided CoMo/Al2O3 catalyst were analyzed by X-ray photoelectron spectroscopy (XPS) and high resolution transmission electron microscopy (HRTEM), respectively. Details of the testing conditions were described previously.7,16
Catalytic activity measurements were carried out in a continuous flow fixed-bed micro-reactor with an inner diameter of 6 mm. The oxidic catalyst (1.0 g) was presulfided in situ at 1.6 Mpa, 360 °C for 2 h, by a stream containing 5 wt% carbon disulfide (CS2) in cyclohexane. Sulfiding feed and H2 flow were 12 mL h−1 and 10.8 L h−1, respectively. After the sulfidation of the catalyst, reacting feed was introduced with a plunger pump into the reactor. The reaction conditions were the following: hydrogen pressure of 1.6 MPa, reaction temperature of 195–285 °C, a liquid feed with a flow rate of 12 mL h−1, and a H2/feed volumetric ratio of 900. Reactions were performed in gas phase. After steady-state conditions were reached, the products were periodically autosampled and analyzed on-line by a gas chromatography equipped with a PONA capillary column (50 m × 0.25 mm × 0.5 μm) and a flame-ionization detector. A feed containing 20 wt% of 1-hexene in heptane was used for the transformation of 1-hexene in the presence of H2. To examine the influence of H2S content on 1-hexene hydroconversion, the olefin concentration was kept constant while hydrogen sulfide content in feed varied from 0 to 1000 μg g−1 S. Thiophene, 2-methylthiophene and 3-methylthiophene were respectively introduced into the feed so as to study the effects of organic sulfur compounds on the transformation of 1-hexene.
The hydrogenation (HYD) and isomerization (ISOM) conversion of 1-hexene, and HDS activity of organic sulfur compounds were calculated as follows:
| | |
HYD (%) = [Hn-hexane in product/H1-hexene in feed] × 100
| (1) |
| | |
ISOM (%) = [H2-,3-hexene in product/H1-hexene in feed] × 100
| (2) |
| | |
HDS (%) = [(Sfeed − Sproduct)/Sfeed] × 100
| (3) |
where
Hn-hexane in product indicates the
n-hexane concentration in the product,
H1-hexene in feed indicates 1-hexene concentration in the feed,
H2-,3-hexene in product indicates the concentration of 2-hexene and 3-hexene in the product, and
Sfeed and
Sproduct indicate the sulfur compound content in the feed and product, respectively.
The pseudo-first-order reaction rate constant of 1-hexene hydrogenation could be determined using the following equation:
| | |
kHYD = −F0/m × ln(1 − HYD)
| (4) |
where
F0 is the reactant molar flow (mol h
−1),
m is the catalyst weight used (g) and HYD is the HYD conversion (%) of 1-hexene.
Results and discussion
Properties of the CoMo/Al2O3 catalyst
Table 1 shows the properties of the support and CoMo catalyst. The alumina support had a BET surface area of 140 m2 g−1 and pore volume of 0.80 mL g−1. The surface acidity of alumina is listed in Table S1.† The cobalt and molybdenum species occupied some part of the pores and hence reduced the surface area and pore volume. The oxidation state CoMo catalyst was sulfided and the sulfidation degree of Mo reached 72.6%. The average slab length and stacking layer number of MoS2 phases were 3.5 nm and 1.7, respectively. It indicated that the MoS2 species were relatively well dispersed on alumina support.
Table 1 Properties of Al2O3 and sulfided CoMo/Al2O3 catalyst
| Sample |
Pore structure |
Mo4+/Mototald (%) |
MoS2 morphology |
| SBETa (m2 g−1) |
Vb (cm3 g−1) |
Dc (nm) |
![[L with combining macron]](https://www.rsc.org/images/entities/i_char_004c_0304.gif) e (nm) e (nm) |
![[N with combining macron]](https://www.rsc.org/images/entities/i_char_004e_0304.gif) e e |
| BET surface area. Pore volume. Average pore size. Sulfidation degree of Mo. Average slab length and stacking layer number obtained by the statistics analysis from HRTEM micrographs. |
| Al2O3 |
140 |
0.80 |
23.0 |
— |
— |
— |
| Sulfided CoMo/Al2O3 |
131 |
0.50 |
15.2 |
72.6 |
3.5 |
1.7 |
Transformation of 1-hexene
Reaction of 1-hexene in the absence of sulfur compounds was performed in order to identify the product distribution of 1-hexene hydroconversion by itself. Under the operating conditions employed herein, the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond of 1-hexene migrated from the terminal position to an internal position. Besides, the hydrogenation of 1-hexene was observed. Reaction products of 1-hexene hydroconversion were n-hexane, trans-2-hexene, cis-2-hexene, trans-3-hexene and cis-3-hexene. The reaction scheme for 1-hexene transformation is shown in Scheme 1. The hydroconversion of 1-hexene over the sulfided catalyst might proceed through two possible reaction pathways: one pathway via 1-hexene double-bond isomerization and direct hydrogenation to n-hexane at the comparable rates, and the other pathway involving 1-hexene double-bond isomerization, followed by hydrogenation to n-hexane.
C double bond of 1-hexene migrated from the terminal position to an internal position. Besides, the hydrogenation of 1-hexene was observed. Reaction products of 1-hexene hydroconversion were n-hexane, trans-2-hexene, cis-2-hexene, trans-3-hexene and cis-3-hexene. The reaction scheme for 1-hexene transformation is shown in Scheme 1. The hydroconversion of 1-hexene over the sulfided catalyst might proceed through two possible reaction pathways: one pathway via 1-hexene double-bond isomerization and direct hydrogenation to n-hexane at the comparable rates, and the other pathway involving 1-hexene double-bond isomerization, followed by hydrogenation to n-hexane.
 |
| | Scheme 1 Reaction scheme for the transformation of 1-hexene. | |
It was accepted that, with the sulfided CoMo/Al2O3 catalyst, olefins were inclined to adsorb on coordinatively unsaturated Mo atoms.24,25 Hydrogen dissociated in the heterolytic way on a sulfur vacancy and an adjacent sulfur anion. Besides, it was evidenced that three hydrogen species were formed on the metal sulfide catalyst: SH groups and two different metal-H linear species.26 It was reported that heterolytic hydrogen participated in the hydrogenation of propene and toluene over sulfides.27,28 Furthermore, the concentration of hydride-type adsorbed species was closely associated with the catalytic activity in hydrogenation reactions.26 According to Breysse et al.29 hydrogen retained on Mo CUS, namely, hydridic Mo species, was responsible for the olefin hydrogenation activity of MoS2. As reported in the literature,30,31 isomerization and hydrogenation of olefin proceeded respectively on mono-hydride (MoH) and di-hydride (MoH2) sites, both of which were formed on the edge of MoS2 in the presence H2. It was generally believed that an activation barrier for dissociative adsorption of H2 existed under reaction conditions. The high temperature and high partial pressure of H2 could facilitate and accelerate the activation of H2 and the creation of sulfur vacancies.32 It was reasonable to expect that, at constant H2 partial pressure, the formation of hydridic Mo species, especially of MoH2 species, could be promoted by increasing temperature. The reaction temperature and reaction atmosphere could influence the state of Mo CUS of the sulfided catalyst and therefore ultimately affect the isomerization and hydrogenation of 1-hexene.
Fig. 1 displays the effect of temperature on the composition of C6 hydrocarbons in the reaction product. It was clearly seen that the total conversion of 1-hexene was above 96%. But the hydrosaturation rate was less than 32% at temperatures lower than 195 °C. This could be explained by the fact that, at lower temperatures, H2 activation and sulfur vacancies formation were relatively slow and the formation of MoH2 species was limited. With the increase of temperature, n-hexane concentration in reaction product increased significantly and internal olefins concentration decreased distinctly. It indicated that increasing temperature favored the hydrogenation of olefins, probably due to the formation of amounts of olefin hydrogenation active sites at elevated temperatures. Therefore, it could be concluded that the double-bond isomerization of 1-hexene proceeded readily at lower temperatures while the hydrogenation of 1-hexene and its isomers predominated at higher temperatures. It was also known that the octane number of a terminal olefin was lower than its corresponding internal olefins.33,34 Hence low reaction temperature contributed to octane boosting and depression of olefin hydrogenation.
 |
| | Fig. 1 Effect of temperature on the composition of C6 hydrocarbons in the reaction product. | |
Effect of H2S on the transformation of 1-hexene
The hydroconversion of 1-hexene in the presence of H2S was investigated. The reactions that 1-hexene underwent were double bond isomerization and hydrogenation. Hexanethiols were not detected by GC analysis. Fig. 2 shows the HYD conversion for 1-hexene as a function of H2S content at different temperatures. It was seen that H2S had little effect on the hydrogenation activity of 1-hexene at temperature higher than 255 °C. However, in the temperature range of 195–255 °C, the inhibiting effect of H2S on the hydrogenation activity was evident. It was shown that, at 240 °C, HYD conversion of 1-hexene increased linearly with decreasing H2S content. More importantly, the saturation rate of 1-hexene increased sharply when H2S content decreased to below 100 μg g−1 S at temperatures below 240 °C. Hence olefins saturated easily in the conventional deep HDS process, probably attributing to the weak inhibition by low-level H2S.
 |
| | Fig. 2 Effect of H2S content on 1-hexene HYD conversion at different temperatures. | |
Tables 2 and 3 display the effect of H2S content on the isomerization and hydrogenation of 1-hexene at 210 °C and at 195 °C, respectively. Regarding the composition of C6 hydrocarbons, unconverted 1-hexene concentration was found to rise distinctly with H2S content increase. This result indicated that the transformation of 1-hexene was retarded by the presence of H2S. It was known that H2S could easily adsorb on Mo CUS of the sulfided hydrotreating catalyst due to its high adsorption heat.35 As reported in the literature,26,27,36,37 H2S and H2 dissociated in the heterolytic way on sulfided Al2O3-supported hydrotreating catalysts. H2S dissociated on a sulfur vacancy of a Mo atom, while the heterolytic dissociation of H2 proceeded on a sulfur vacancy and an adjacent sulfur anion. It was reasonable to assume that the presence of H2S could impede the dissociation of H2. Therefore, the inhibition of H2S on 1-hexene transformation could be attributed to the competition adsorption between H2S and H2 on Mo CUS.
Table 2 Effect of H2S content on the isomerization and hydrogenation of 1-hexene at 210 °C
| H2S content, μg g−1 S |
Composition in C6 hydrocarbons (composition in olefins), % |
| nC6 |
1-C6 |
2-C6 |
3-C6 |
2-C6 and 3-C6 |
| 0 |
85.0 |
0.8 (5.1) |
10.3 (68.6) |
3.9 (26.3) |
14.2 |
| 100 |
24.0 |
4.0 (5.2) |
51.8 (68.2) |
20.2 (26.6) |
72.0 |
| 250 |
18.0 |
4.2 (5.1) |
56.0 (68.3) |
21.8 (26.6) |
77.8 |
| 500 |
14.2 |
4.4 (5.1) |
58.7 (68.5) |
22.7 (26.4) |
81.4 |
| 1000 |
9.7 |
4.8 (5.3) |
63.5 (70.4) |
22.0 (24.3) |
85.5 |
Table 3 Effect of H2S content on the isomerization and hydrogenation of 1-hexene at 195 °C
| H2S content, μg g−1 |
Composition in C6 hydrocarbons (composition in olefins), % |
| nC6 |
1-C6 |
2-C6 |
3-C6 |
2-C6 and 3-C6 |
| 0 |
31.4 |
3.2 (4.6) |
47.4 (69.1) |
18.0 (26.3) |
65.4 |
| 100 |
9.4 |
4.4 (4.8) |
63.0 (69.6) |
23.1 (25.5) |
86.2 |
| 250 |
7.4 |
4.9 (5.3) |
67.5 (72.9) |
20.2 (21.8) |
87.7 |
| 500 |
6.8 |
7.2 (7.7) |
71.4 (76.6) |
14.7 (15.7) |
86.0 |
| 1000 |
5.9 |
17.2 (18.2) |
68.5 (72.8) |
8.5 (9.0) |
76.9 |
With respect to the composition of C6 hydrocarbons (given in Tables 2 and 3), the concentration of n-hexane decreased with H2S content increase. As mentioned above, the active sites for olefin hydrogenation reaction were MoH2 species, which were created by H2 dissociative adsorption on Mo CUS. Thus the inhibiting effect of H2S on olefin hydrogenation might be explained by the fact that the dissociative adsorption of H2S could decrease the amount of highly coordinatively unsaturated Mo atoms and be ultimately detrimental to the formation MoH2.
As presented in Table 2, at 210 °C, the composition of C6 olefins remained approximately constant when H2S content increased from 0 to 500 μg g−1 S. Besides, it was seen that only a small amount of 3-hexene was converted into 2-hexene at H2S level of 1000 μg g−1 S. These results indicated that, at 210 °C, the isomerization reaction of 1-hexene almost approached chemical equilibrium and H2S had limited impact on 1-hexene isomerization. At 195 °C, as given in Table 3, the composition of olefins changed greatly with H2S content increase. The concentration of 1-hexene rised from 4.6% to 18.2% and 3-hexene was constantly converted into 2-hexene when increasing H2S content from 0 to 1000 μg g−1 S. It suggested that the isomerization reaction of 1-hexene was impeded by H2S at 195 °C.
It was noted that ISOM products distinctly increased in the presence of H2S, due to the strong inhibition of H2S on the hydrogenation of 1-hexene and its isomers. Additionally, the inhibiting effect of H2S on both isomerization and hydrogenation reactions resulted in the decline in 1-hexene HYD conversion.
Effects of organic sulfur compounds on the transformation of 1-hexene
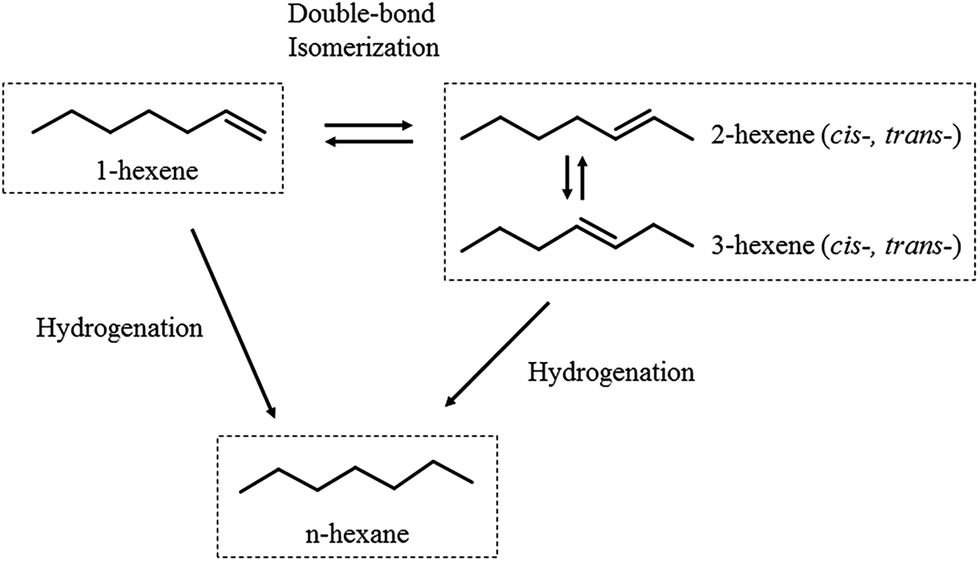
Four mixed feeds with 1-hexene (20 wt%) and 500 μg g−1 S of respectively 2MT, 3MT, T and H2S were tested under the same reaction conditions. Fig. 3 shows the HYD conversion of 1-hexene in the presence of organic sulfides over the temperature range 195–285 °C. It could be seen that organic sulfur compounds had no effect on the hydrosaturation of 1-hexene at temperature higher than 255 °C. But the HYD reaction was retarded to the same extent by three organic sulfur compounds and H2S at temperatures between 240 °C and 255 °C. It could be easily explained by the fact that T, 2MT and 3MT were completely desulfurized to form H2S in this condition (given in Fig. 4). Especially, in the temperature range of 195–240 °C, the impact of inhibition on 1-hexene HYD activity followed the order H2S > thiophene > 3-methylthiophene > 2-methylthiophene. As shown in Fig. 4, three organic sulfides were partly converted to H2S in the reaction system at the temperature of below 240 °C. Hence, the inhibiting effect on 1-hexene HYD reaction may result from organic sulfur compound or H2S generated by organic sulfide desulfurization.
 |
| | Fig. 3 Effects of organic sulfur compounds on the HYD conversion of 1-hexene. | |
 |
| | Fig. 4 Changes in HDS activities as a function of reaction temperature. | |
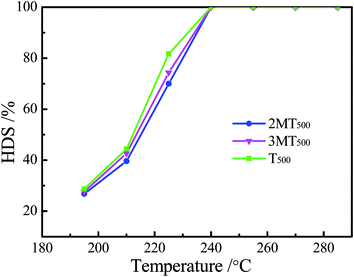
One feed containing 20 wt% of 1-hexene and 1000 μg g−1 S of thiophene, and another feed containing 20 wt% of 1-hexene, 500 μg g−1 S of thiophene and 500 μg g−1 S of H2S were carried out under the same operating conditions as mentioned before. The obtained results were compared with experiment data taken from Fig. 3 and 4 to figure out from which the indeed inhibition of 1-hexene HYD reaction originated, organic sulfur compound or H2S generated by hydrodesulfurization. Fig. 5 presents HYD reaction apparent kinetic constant versus sulfur compound content at the inlet of reactor. It was clearly seen that the inhibition impact of organic sulfide was slightly lower than that of H2S on HYD reaction. Especially, a different trend in the same HYD reaction apparent kinetic constant as a function of H2S content at reactor outlet is shown in Fig. 6. The influence of organic sulfur compound on 1-hexene HYD reaction could be explained by the fact that H2S produced by organic sulfides hydrodesulfurization inhibited the apparent kinetic constant of HYD reaction. Therefore, the inhibiting effect of organic sulfur compound on 1-hexene HYD reaction could be attributed to H2S generated by hydrodesulfurization instead of organic sulfur compound. A similar result was also reported by Santos et al.38
 |
| | Fig. 5 HYD reaction apparent kinetic constant versus sulfur compound content at the inlet of reactor. | |
 |
| | Fig. 6 HYD reaction apparent kinetic constant versus H2S content at the outlet of reactor. | |
Fig. 7 displays the effects of organic sulfur compounds on 1-hexene ISOM conversion at different temperatures. A distinct increase in ISOM conversion was observed in the presence of sulfur compounds. Besides, 1-hexene ISOM conversion decreased with increasing the reaction temperature. Table 4 shows the effects of organic sulfur compounds on the isomerization reaction of 1-hexene at 210 °C and 195 °C. It could be seen that the presence of 500 μg g−1 S of organic sulfides seldom changed the composition of C6 olefins. This suggested that, at 210 °C and 195 °C, ISOM reaction of 1-hexene approached chemical equilibrium rapidly and seemed unaffected by organic sulfides. As presented in Fig. 4, the CoMo catalyst exhibited low HDS activity at 195 °C. Only small amount of H2S were produced by organic sulfides hydrodesulfurization. Thus organic sulfur compounds had little impact on the isomerization reaction of 1-hexene. But more isomerization products were obtained in the presence of sulfur compounds, probably attributable to the inhibition of sulfur compounds on the hydrogenation of 1-hexene and its isomers.
 |
| | Fig. 7 Effects of organic sulfur compounds on 1-hexene ISOM conversion at different temperatures. | |
Table 4 Effects of organic sulfur compounds on the isomerization of 1-hexene at 210 °C and 195 °C
| Sulfide |
Composition in C6 olefins at 210 °C, % |
Composition in C6 olefins at 195 °C, % |
| 1-C6 |
2-C6 |
3-C6 |
1-C6 |
2-C6 |
3-C6 |
| Pure 1-hexene. |
| S0a |
5.1 |
68.6 |
26.3 |
4.6 |
69.1 |
26.3 |
| 2MT500 |
5.2 |
68.3 |
26.5 |
4.8 |
68.8 |
26.4 |
| 3MT500 |
5.2 |
68.3 |
26.5 |
4.8 |
68.7 |
26.5 |
| T500 |
5.2 |
68.3 |
26.5 |
4.8 |
68.8 |
26.4 |
| H2S500 |
5.1 |
68.5 |
26.4 |
7.7 |
76.6 |
15.7 |
Conclusions
Hydrogenation and isomerization reactions of 1-hexene were observed over a sulfided CoMo/Al2O3 catalyst. The double-bond isomerization of 1-hexene proceeded readily at lower temperatures while the hydrogenation of 1-hexene and its isomers predominated at higher temperatures. The inhibiting effect of H2S on both isomerization and hydrogenation reactions resulted in the decline in 1-hexene HYD conversion. The saturation rate of 1-hexene increased sharply when H2S content decreased to below 100 μg g−1 S. It was also found that thiophene, 2-methylthiophene and 3-methylthiophene strongly inhibited the hydrogenation of 1-hexene. Furthermore, the inhibiting effect on 1-hexene HYD reaction originated from H2S generated by organic sulfide desulfurization. More isomerization products were obtained in the presence of sulfur compounds, probably attributable to the inhibition of sulfur compounds on the hydrogenation of 1-hexene and its isomers.
Acknowledgements
Financial support from the National Key Basic Research and Development Program of China (973 program, No. 2012 CB224802) is gratefully acknowledged.
References
- S. Brunet, D. Mey, G. Pérot, C. Bouchy and F. Diehl, Appl. Catal., A, 2005, 278, 143–172 CrossRef CAS.
- A. Daudin, A. F. Lamic, G. Perot, S. Brunet, P. Raybaud and C. Bouchy, Catal. Today, 2008, 130, 221–230 CrossRef CAS.
- C. Song, Catal. Today, 2003, 86, 211–263 CrossRef CAS.
- A. Pashigreva, G. Bukhtiyarova, O. Klimov, Y. A. Chesalov, G. Litvak and A. Noskov, Catal. Today, 2010, 149, 19–27 CrossRef CAS.
- Y. Okamoto, A. Kato, N. Rinaldi, T. Fujikawa, H. Koshika, I. Hiromitsu and T. Kubota, J. Catal., 2009, 265, 216–228 CrossRef CAS.
- M. Breysse, C. Geantet, P. Afanasiev, J. Blanchard and M. Vrinat, Catal. Today, 2008, 130, 3–13 CrossRef CAS.
- M. Li, H. Li, F. Jiang, Y. Chu and H. Nie, Fuel, 2009, 88, 1281–1285 CrossRef CAS.
- P. Nikulshin, D. Ishutenko, A. Mozhaev, K. Maslakov and A. Pimerzin, J. Catal., 2014, 312, 152–169 CrossRef CAS.
- H. Ge, X.-D. Wen, M. A. Ramos, R. R. Chianelli, S. Wang, J. Wang, Z. Qin, Z. Lyu and X. Li, ACS Catal., 2014, 4, 2556–2565 CrossRef CAS.
- E. Hensen, P. Kooyman, Y. Van der Meer, A. Van der Kraan, V. De Beer, J. Van Veen and R. Van Santen, J. Catal., 2001, 199, 224–235 CrossRef CAS.
- H. Topsøe, B. Hinnemann, J. K. Nørskov, J. V. Lauritsen, F. Besenbacher, P. L. Hansen, G. Hytoft, R. G. Egeberg and K. G. Knudsen, Catal. Today, 2005, 107, 12–22 CrossRef.
- F. Besenbacher, M. Brorson, B. Clausen, S. Helveg, B. Hinnemann, J. Kibsgaard, J. V. Lauritsen, P. G. Moses, J. K. Nørskov and H. Topsøe, Catal. Today, 2008, 130, 86–96 CrossRef CAS.
- S. Eijsbouts, Appl. Catal., A, 1997, 158, 53–92 CrossRef CAS.
- H. Topsøe, Appl. Catal., A, 2007, 322, 3–8 CrossRef.
- J. Chen, F. Maugé, J. El Fallah and L. Oliviero, J. Catal., 2014, 320, 170–179 CrossRef CAS.
- M. Li, H. Li, F. Jiang, Y. Chu and H. Nie, Catal. Today, 2010, 149, 35–39 CrossRef CAS.
- P. Leflaive, J. Lemberton, G. Perot, C. Mirgain, J. Carriat and J. Colin, Appl. Catal., A, 2002, 227, 201–215 CrossRef CAS.
- M. Toba, Y. Miki, T. Matsui, M. Harada and Y. Yoshimura, Appl. Catal., B, 2007, 70, 542–547 CrossRef CAS.
- G. Shi, H. Zhao, L. Song and J. Shen, Energy Fuels, 2008, 22, 2450–2454 CrossRef CAS.
- Y. Shi, D. Li, Y. Xi and H. Wu, Pet. Process. Petrochem., 2010, 41, 28–31 CAS.
- Y. Xi, X. Gao, M. Li and H. Nie, Pet. Process. Petrochem., 2009, 40, 1–4 CAS.
- A.-F. Lamic, A. Daudin, S. Brunet, C. Legens, C. Bouchy and E. Devers, Appl. Catal., A, 2008, 344, 198–204 CrossRef CAS.
- M. Toba, Y. Miki, Y. Kanda, T. Matsui, M. Harada and Y. Yoshimura, Catal. Today, 2005, 104, 64–69 CrossRef CAS.
- T. Mochizuki, H. Itou, M. Toba, Y. Miki and Y. Yoshimura, Energy Fuels, 2008, 22, 1456–1462 CrossRef CAS.
- F. Di-Grégorio, V. Keller, T. Di-Costanzo, J. Vignes, D. Michel and G. Maire, Appl. Catal., A, 2001, 218, 13–24 CrossRef.
- H. Jobic, G. Clugnet, M. Lacroix, S. Yuan, C. Mirodatos and M. Breysse, J. Am. Chem. Soc., 1993, 115, 3654–3657 CrossRef CAS.
- M. Brémaud, L. Vivier, G. Pérot, V. Harlé and C. Bouchy, Appl. Catal., A, 2005, 289, 44–50 CrossRef.
- S. Kasztelan and D. Guillaume, Ind. Eng. Chem. Res., 1994, 33, 203–210 CrossRef CAS.
- M. Breysse, E. Furimsky, S. Kasztelan, M. Lacroix and G. Perot, Catal. Rev., 2002, 44, 651–735 CAS.
- K.-I. Tanaka, Appl. Catal., A, 1999, 188, 37–52 CrossRef CAS.
- K.-I. Tanaka and N. Takehiro, J. Mol. Catal. A: Chem., 1999, 141, 39–55 CrossRef CAS.
- J. Lauritsen, M. Nyberg, J. K. Nørskov, B. Clausen, H. Topsøe, E. Lægsgaard and F. Besenbacher, J. Catal., 2004, 224, 94–106 CrossRef CAS.
- D. Mey, S. Brunet, C. Canaff, F. Maugé, C. Bouchy and F. Diehl, J. Catal., 2004, 227, 436–447 CrossRef CAS.
- C. Yin, R. Zhao and C. Liu, Fuel, 2005, 84, 701–706 CrossRef CAS.
- B. Liu, Y. Chai, Y. Li, A. Wang, Y. Liu and C. Liu, Fuel, 2014, 123, 43–51 CrossRef CAS.
- C. Thomas, L. Vivier, A. Travert, F. Maugé, S. Kasztelan and G. Pérot, J. Catal., 1998, 179, 495–502 CrossRef CAS.
- C. Thomas, L. Vivier, J. Lemberton, S. Kasztelan and G. Pérot, J. Catal., 1997, 167, 1–11 CrossRef CAS.
- N. Dos Santos, H. Dulot, N. Marchal and M. Vrinat, Appl. Catal., A, 2009, 352, 114–123 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra27583k |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.