
Core–shell SrTiO3:Yb3+,Er3+@mSiO2 nanoparticles for controlled and monitored doxorubicin delivery†
Binbin Lia,
Heng Liua,
Chuanbin Sunb,
Zeeshan Ahmadc,
Zhaohui Rena,
Xiang Li*a and
Gaorong Han*a
aState Key Laboratory of Silicon Materials, School of Materials Science and Engineering, Zhejiang University, Hangzhou 310027, PR China. E-mail: xiang.li@zju.edu.cn; hgr@zju.edu.cn
bEye Center, Second Affiliated Hospital, School of Medicine, Zhejiang University, PR China
cLeicester School of Pharmacy, De Montfort University, Leicester, LE1 9BH, UK
First published on 3rd March 2016
Abstract
The investigation of nano-carriers with controllable and trackable drug release kinetics has attracted worldwide attention for new tumor theranostic strategies with catabatic side effects. Herein, a range of monodispersed core–shell structured photoluminescent SrTiO3:Yb3+,Er3+@mSiO2 nanoparticles were designed and synthesized. The surfactant cetyltrimethylammonium bromide (CTAB) was used to vary the microstructure of the mesoporous SiO2 shell. The specific surface area and pore volume increase proportionally with the content of CTAB. Consequently, the doxorubicin (DOX) loading capacity increases, and the drug release kinetics possesses a sustained behaviour. More importantly, the DOX release kinetics was found to correspond well to the evolution of the up-conversion luminescence (UCL) phenomenon. More rapid drug release induces more rapid photoluminescence enhancement, and vice versa. This study has therefore been anticipated to suggest another promising multifunctional drug delivery platform for advanced chemotherapies.
1. Introduction
Multifunctional drug delivery systems (DDS) with controlled drug release behaviour have received worldwide attention for tumor diagnosis and therapy. DDS with sustained drug release properties, especially at the nanoscale, can deliver therapeutic drugs to the targeted sites at an expected concentration over a prolonged period, and thus improve the therapeutic effectiveness.1 Generally, an ideal drug delivery system is expected to transport the desired drug molecules to the targeted tissues or cells, and release in a controlled manner. To achieve this goal, not only the drug release efficiency and the selection of therapeutic means are of particular interest, the monitoring and tracking of drug releasing phenomenon in an external and in situ way during the therapeutic process is equally vital.Light-responsive nanomaterials have been extensively investigated for cell imaging and drug delivery.2 Conventional luminescent materials, such as fluorescent proteins, quantum dots and organic dyes, have suffered a range of inherent disadvantages, such as DNA damage and cell death due to long-term exposure to high energy excitation, low signal-to-noise ratio (SNR) caused by significant auto-fluorescence from the biological tissues, and low penetration depth in the biological tissues.3 In the contrast, upconversion nanoparticles (UCNPs), which emit the spectra from ultraviolet (UV) to near infrared reflection (NIR) due to the absorption of NIR photons, have emerged as promising candidate with weak auto-fluorescence from biological substances, and low cytotoxicity.4 More importantly, the excitation and emission wavelengths of upconversion luminescence (UCL) process can be manipulated to fit the “optical transmission window” in which living tissues are maximally transparent.5 Among rare earth ions doped into UCNPs, Er3+ ion is the most popular activator which possesses ladder-like arrangement energy levels and strong excited state absorption at 980 nm.6 Generally, the content of activators is relatively low (usually <2 mol%), minimizing any cross-relaxation energy loss. Yb3+ acts as a sensitizer to enhance the UCL efficiency for larger absorption cross-section at ∼980 nm than other lanthanide ions.7
Recently, a range of rare earth doped UCNPs have been investigated for biomedical applications, especially as the fluorescence labels and delivery of drug molecules. For example, a model drug, ibuprofen, was delivered via nanofibers containing mesoporous silica coated β-NaYF4:Yb,Er UCNPs.8 Ibuprofen has high vibration energy, and thus it significantly quenches the upconversion luminescence of the nanocomposite when loaded. The subsequent drug-release process was followed by the recovery of upconversion luminescence emission intensity, indicating the potential of this method for tracking the cumulative release of drugs loaded. UCNPs have also been employed to deliver nucleic acids in gene therapy. The feasibility of using silica-coated β-NaYF4:Yb,Er UCNPs to track the delivery of siRNA into cells was demonstrated using a UCNPs-based luminescence resonance energy transfer (LRET) system for studying the intracellular uptake, drug release, and bio-stability of UCNPs-bound siRNA in live cells.9 When amino-group-modified UCNP was bound with BOBO-3-stained siRNA, energy was efficiently transferred from UCNPs to BOBO-3 to generate characteristic BOBO emissions at 602 nm under excitation at the 980 nm spectrum. Once siRNA molecules are detached from UCNPs, the LRET process is inhibited. The study showed that siRNA was gradually released from the UCNPs surface into the cytoplasm over 24 h, correlating well with further fluorescent co-localization imaging validation. However, some studies also reported that many nanomaterials can induce cytotoxic effect when cells are long-time exposure to high concentrations of nanoparticles (e.g. NaYF4, GdF3) due to its incompatible chemical compositions to the natural tissue and nano-toxicity effect.10 The research in the suitable host matrix for UCNPs with biocompatibility and lower cytotoxity for tumor diagnosis and therapy is still been endeavored.
Strontium titanate (SrTiO3, STO) has been used as a coating material for orthopedic applications. The strontium element was found to play an inhibiting role in bone resorption, and stimulate the new bone formation.11 The in vitro study has confirmed that the crystalline SrTiO3 possesses good biocompatibility with profound biominerallization abilities.12 Recently, it was found that SrTiO3 polyhedral nanocrystals induce the facet-specific assembly of proteins, such as albumin, immunoglobulin and protamine.13 Furthermore, Er can be feasibly doped and caged within the lattice of SrTiO3 to avoid possible lanthanide element releasing due to its perovskite crystal structure and the dimensional matching between Er3+ and Sr2+ ions.14 Therefore, SrTiO3 is rather suitable as host matrix of rare earth ions for light-responsive DDS, but the drug delivery systems based on this system is still lacking.
In this study, a series of SrTiO3:Yb3+,Er3+ (STO:Yb3+,Er3+) nanoparticles coated with mesoporous silica shell (STO:Yb3+,Er3+@mSiO2) have been designed and synthesized as a novel drug delivery platform for the first time. The microstructure of the mSiO2 shell was manipulated by using the surfactant cetyltrimethylammonium bromide (CTAB). Doxorubicin (DOX), a well-documented anti-cancer agent, was used as the model drug. The effect of the shell microstructure on the DOX loading and releasing behaviors were uncovered in-depth. More importantly, the variation of photoluminescent phenomenon during the DOX release progress was investigated. Such photoluminescent STO:Yb3+,Er3+@mSiO2 nanoparticles with controlled core–shell structure have been considered as a new drug delivery platform for the future tumor diagnosis and therapy.
2. Materials and methods
2.1. Materials
Strontium nitrate (Sr(NO3)2, A.R.), sodium hydroxide (NaOH, A.R.), titanium chloride (TiCl3, A.R.), cyclohexane (C6H12, A.R.), phosphate-buffered saline (PBS, pH = 7.4) and tetraethylorthosilicate (TEOS, C.P.) were purchased from Sinopharm Chemical Reagent Co., Ltd. Oleic acid (C18H34O2, A.R.), sodium oleate (C17H33CO2Na, C.P.), cetyltrimethylammonium bromide (CTAB, 99%), doxorubicin (DOX, 98%), erbium nitratepentahydrate (Er(NO3)3·5H2O, 99.9%) and ytterbium nitratepentahydrate (Yb(NO3)3·5H2O, 99.9%) were purchased from Aladdin. All reagents were used without further purification.2.2. The synthesis of STO:Yb3+,Er3+ nanoparticles (NPs)
The STO:Yb3+,Er3+ NPs were synthesized using bi-metal and rare earth nitrate Re(NO3)3 (RE = 18% Yb + 2% Er) precursors by liquid–solid-solution (LSS) method.15 In a typical procedure, a mixed solution of 4 mmol TiCl3 and 10 mL ethanol were added dropwise to 10 mL NaOH solution (0.7 g mL−1) to prepare a milk-like suspension. 2 mL oleic acid and 1.6 g sodium oleate were sequentially added to the solution. Sr(NO3)2, Er(NO3)3·5H2O and Yb(NO3)3·5H2O (4 mmol in total, of which 80 mol% Sr, 18 mol% Yb and 2 mol% Er) were added dropwise into 5 mL deionized water. The mixture was then transferred into a 50 mL Teflon-lined vessel and added with deionized water to prepare 40 mL solution. After being sealed in an autoclave, the solution was heat-treated at 180 °C for 8 h, and cooled to the ambient temperature. The resulting white precipitates were further purified using cyclohexane and ethanol. The final samples were collected by means of centrifugation, washed with ethanol, and then dispersed in cyclohexane at a concentration of 10 mg mL−1.2.3. Surface modification of STO:Yb3+,Er3+ nanoparticles with mSiO2 shell
A typical forming process of mesoporous SiO2 shell, as demonstrated in Fig. 1, was performed by the self-assembly process of the CTAB and TEOS.16 2 mL STO:Yb3+,Er3+ NPs suspension prepared was added into 5 mL aqueous CTAB solution (0.0275, 0.055 and 0.0825 M) with stirring and sonication vigorously NPs/CTAB solution. The resulting solution was added to a mixture of water (45 mL), NaOH solution (0.3 mL, 2 M) and 2.2 mmol for 30 min.17 Subsequently, the mixture was heated up to 70 °C under stirring to evaporate the cyclohexane, resulting in a tetraethylorthosilicate (TEOS) in sequence under stirring. The synthesized samples were washed 3 times with ethanol, and collected. The powder prepared was then dried at 80 °C overnight and calcined at 550 °C for 5 h. The obtained samples are denoted as STO:Yb3+,Er3+@xmSiO2, where x (0.5, 1.0 and 1.5) represents the concentration of CTAB (0.0275, 0.055 and 0.0825 M, respectively). | ||
| Fig. 1 Schematic illustration of the synthetic route to STO:Yb3+,Er3+@mSiO2 nanoparticles. | ||
STO:Yb3+,Er3+ NPs and STO:Yb3+,Er3+@mSiO2 NPs were characterized using field emission scanning electron microscopy (FESEM, Hitachi SU-70, Japan) and high-resolution transmission electron microscopy (HRTEM, Tecnai F20, FEI, USA), X-ray diffraction (XRD, ARLXTRA, Thermo, Switzerland, Cu Kα) and Fourier Transform Infrared spectroscopy (FTIR Tensor 27, Bruker, Germany) with the KBr pellet. The particle dimension was quantified by measuring 100 particles stochastically. The up-conversion (UC) emission spectra were recorded on an F-7000 spectrophotometer (Edinburgh FLSP920), equipped with a 980 nm laser as the excitation source and detected from 500 to 750 nm.
2.4. In vitro cytotoxicity assessment
The cellular toxicity of the as synthesized nanoparticles was determined by means of a standard MTT cell assay. L929 fibroblast cells were seeded in a 96-well plate at a density of 8000 cells per well in the medium, and then cultured at 37 °C under 5% CO2 for 24 h to allow the cells to attach to the wells. Subsequently, STO:Yb3+,Er3+@mSiO2 nanoparticles were added to the medium, and the cells were incubated in 5% CO2 at 37 °C for 48 h. Different concentrations (15, 30, 60, 125 μg mL−1) were used. After incubation, the medium was removed, and 100 μL of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) solution (diluted in a culture media with a final concentration of 0.5 mg mL−1) was added and incubated for another 4 h. The medium was then replaced with 100 μL of dimethyl sulfoxide (DMSO) per well, and the absorbance was monitored using a microplate reader (Bio-TekELx800, USA) at the wavelength of 490 nm.2.5. Drug loading and release
Doxorubicin (DOX) was used as model drug to investigate the drug loading and releasing properties. It was firstly dissolved in phosphate buffered saline (PBS) solution to prepare a 0.8 mg mL−1 DOX–PBS solution. 20 mg STO:Yb3+,Er3+@mSiO2 NPs were immersed in 10 mL DOX–PBS solution with stirring for 48 h at room temperature to reach the equilibrium state. Subsequently, the NPs were collected by centrifuging at 8000 rpm for 15 minutes, and washed with deionized water to remove the unloaded DOX. Finally, the samples were dried at 37 °C overnight.18 To determine the amount of DOX content loaded into the NPs, thermogravimetry scanning calorimetry (TG, DSCQ1000, AT, USA) was employed to measure the weight loss of as-prepared product before and after loading with a heating rate of 10 °C min−1 from 25 °C to 650 °C.5,19For DOX release examination, 10 mg of the DOX-loaded STO:Yb3+,Er3+@mSiO2 NPs were soaked in 3 mL fresh PBS at 37 °C. At predetermined time intervals, 1 mL media was taken out and replaced with an equal volume of fresh PBS. The amount of DOX released in the supernatant solutions was measured by UV-vis spectrophotometer (TU-1810, China) at a wavelength of 480 nm. Tests were performed in three replicates.
3. Results and discussions
3.1. Synthesis of STO:Yb3+,Er3+@mSiO2 nanoparticles
 | ||
| Fig. 2 (a) SEM image, (b) TEM image (the inset are HRTEM and SAED images), (c) size distribution and (d) Up-Conversion Luminescent (UCL) spectra of the as-synthesized STO:Yb3+,Er3+ nanoparticles. | ||
Energy Dispersive X-ray (EDX) examination was carried to confirm the element composition of the particles synthesized. Sr![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Yb:Er molar ratio of the particles was estimated to be 8.32:1.94:0.19 which is quite close to the value set (0.8:0.18:0.02) during the experiment (Fig. S1†), implying the successful doping of RE atoms. One notable fact is that the upper doping concentration limit for keeping single phase SrTiO3 was estimated to be 6 mol%, above that multiphase can be induced, for STO:Yb3+,Er3+ nanoparticles synthesized by citric assisted route.21 A possible reason for such different doping concentration could be the highly varied reaction conditions applied in two synthesis approaches. The high pressure and relatively low temperature, induced by LSS method used, may facilitate the atomic diffusion and crystallization of STO:Yb3+,Er3+ particles. The findings agrees well with the study carried out by Yoshiaki Kinemuchi et al., which demonstrated that Sr0.8Y0.2TiO3 nanoparticles, synthesized following the same approach, maintained the pure cubic SrTiO3 phase after annealing at 500 °C.22
:Yb:Er molar ratio of the particles was estimated to be 8.32:1.94:0.19 which is quite close to the value set (0.8:0.18:0.02) during the experiment (Fig. S1†), implying the successful doping of RE atoms. One notable fact is that the upper doping concentration limit for keeping single phase SrTiO3 was estimated to be 6 mol%, above that multiphase can be induced, for STO:Yb3+,Er3+ nanoparticles synthesized by citric assisted route.21 A possible reason for such different doping concentration could be the highly varied reaction conditions applied in two synthesis approaches. The high pressure and relatively low temperature, induced by LSS method used, may facilitate the atomic diffusion and crystallization of STO:Yb3+,Er3+ particles. The findings agrees well with the study carried out by Yoshiaki Kinemuchi et al., which demonstrated that Sr0.8Y0.2TiO3 nanoparticles, synthesized following the same approach, maintained the pure cubic SrTiO3 phase after annealing at 500 °C.22
Another key feature of the synthesized NPs is demonstrated in Fig. 2d. STO:Yb3+,Er3+ NPs exhibit significant up-conversion luminescence phenomenon. Under the excitation of 980 nm spectrum, STO:Yb3+,Er3+ NPs emit visible green and red sprectra. Such UC bands are ascribed to the transitions 2H11/2–4I15/2 (521 nm), 4S3/2–4I15/2 (541 nm), and 4F9/2–4I15/2 (662 nm) due to the Er3+ ions doped.23 Yb3+ acts as the sensitizer for the large cross section. The mechanism of Yb3+, Er3+ cooperation is demonstrated in Fig. S2.† In an initial energy transfer, Yb3+ions are excited from 2F7/2 to 2F5/2 level by 980 nm laser, and transfer its energy to Er3+ (4I11/2). Just as the electron stays on the 4I11/2 level, a second 980 nm photon excites Yb3+ ions, and then the energy is transferred to Er3+, resulting in the electron population on higher 4F7/2 energetic state of the Er3+ ions. The Er3+ ion relaxes nonradiatively by a multi-phonon relaxation process to the 2H11/2 and 4S3/2 levels, and consequently the green 2H11/2–4I15/2 and 4S3/2–4I15/2 emissions occur. Alternatively, the electron can further relax and populate the 4F9/2 level, resulting in the occurrence of dominant red 4F9/2 → 4I15/2 emission.24
 | ||
| Fig. 3 (a) TEM image of STO:Yb3+,Er3+@mSiO2 core–shell structure; (b) XRD patterns of STO:Yb3+,Er3+ with and without mSiO2 shell; (c) schematic illustration of mesoporous silica shell formation; and (d) UCL spectra before and after formation of mesoporous SiO2 shell. | ||
The influence of the mesoporous SiO2 shell to the photoluminescent properties was further studied. As shown in Fig. 3d, the luminescent intensity increases dramatically after coating of mesoporous SiO2 layer. This is because the organic groups in OA with high vibration frequencies from 2800 to 3000 cm−1 quenched the emission of Er3+ in the STO:Yb3+,Er3+. After annealed at 550 °C for 5 h, the organic groups were burned off, inducing the increased intensity with higher magnitude both red and green emission.25
FTIR analysis was carried out to verify the forming mechanism of the STO:Yb3+,Er3+@mSiO2 core–shell nanoparticles. As shown in Fig. 4, the FT-IR spectrum of the as-prepared OA capped STO:Yb3+,Er3+ nanoparticles exhibits the transmittance peaks at 2924 cm−1 and 2853 cm−1 which are attributed to the antisymmetric and symmetric methylene stretches (νas(CH2), νs(CH2)) of the oleic acid (OA) molecules. The peaks appearing between 1400–1600 cm−1 correspond to the ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) stretch, as reported elsewhere.26 When the nanoparticles are coated with a layer of TEOS and CTAB surfactant, the peaks at 2924 cm−1 and 2853 cm−1 became stronger, and meanwhile the peaks appearing between 1400 cm−1 and 1600 cm−1 weakened. Then, a silica precursor tetraethoxysilane (TEOS) hydrolyzed, condensed, and assembled with surfactant micelles to produce CTAB/silica complexes.27 Further, after heating at 550 °C, the FT-IR spectrum of STO:Yb3+,Er3+@1.0CTAB/SiO2 presented the absorption bands at 1086 cm−1, 798 cm−1 and 460 cm−1, indicating the Si–O–Si framework was formed. The transmittance band at 958 cm−1 is attributed to the Si–OH asymmetric stretching.28 Meanwhile, as the organic molecules were decomposed during heating procedure, the absorption bands of –CH2– and CO disappeared, and thus the photoluminescent intensity of the nanoparticles were dramatically enhanced. The similar phenomena were observed from STO:Yb3+,Er3+@0.5mSiO2 and STO:Yb3+,Er3+@1.5mSiO2 nanoparticles synthesized, as shown in the ESI (Fig. S3†).
O) stretch, as reported elsewhere.26 When the nanoparticles are coated with a layer of TEOS and CTAB surfactant, the peaks at 2924 cm−1 and 2853 cm−1 became stronger, and meanwhile the peaks appearing between 1400 cm−1 and 1600 cm−1 weakened. Then, a silica precursor tetraethoxysilane (TEOS) hydrolyzed, condensed, and assembled with surfactant micelles to produce CTAB/silica complexes.27 Further, after heating at 550 °C, the FT-IR spectrum of STO:Yb3+,Er3+@1.0CTAB/SiO2 presented the absorption bands at 1086 cm−1, 798 cm−1 and 460 cm−1, indicating the Si–O–Si framework was formed. The transmittance band at 958 cm−1 is attributed to the Si–OH asymmetric stretching.28 Meanwhile, as the organic molecules were decomposed during heating procedure, the absorption bands of –CH2– and CO disappeared, and thus the photoluminescent intensity of the nanoparticles were dramatically enhanced. The similar phenomena were observed from STO:Yb3+,Er3+@0.5mSiO2 and STO:Yb3+,Er3+@1.5mSiO2 nanoparticles synthesized, as shown in the ESI (Fig. S3†).
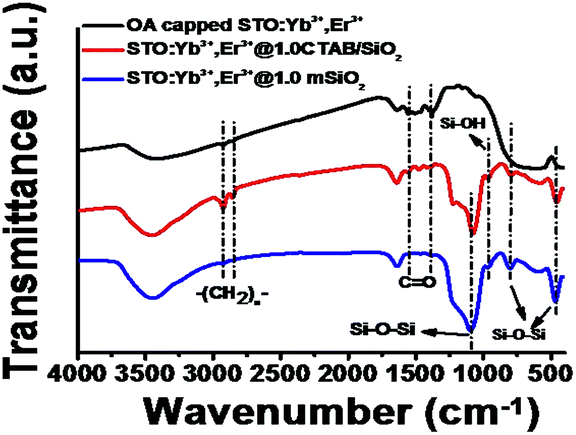 | ||
| Fig. 4 FTIR spectra of OA capped STO:Yb3+, Er3+ nanoparticles, STO:Yb3+,Er3+ nanoparticles coated with a layer of TEOS and CTAB surfactant, and STO:Yb3+,Er3+@1.0mSiO2 nanoparticles. | ||
The physicochemical properties of STO:Yb3+,Er3+@mSiO2 NPs can be varied by incorporating different contents of CTAB (e.g. STO:Yb3+,Er3+@xmSiO2) during the synthesis procedure. The obtained samples are denoted as STO:Yb3+,Er3+@xmSiO2, where x (0.5, 1.0 and 1.5) represents the varied concentration of CTAB concentrations (0.0275, 0.055 and 0.0825 M). The specific surface area, pore size distribution and pore volume of such three NPs were examined using N2 adsorption–desorption method. As shown in Fig. 5, all resulting isotherms display typical characteristics of (type IV) mesoporous materials. The pore size from all three samples, calculated from the desorption branch of the nitrogen isotherm using the Barrett–Joyner–Halenda (BJH) method, is ∼2.5 nm with narrow pore size distribution.29 As summarized in Table 1, when the CTAB concentration was set at 0.5 m, the specific surface area and pore volume are ∼90 m2 g−1 and 0.25 cm3 g−1, respectively. With the increased CTAB concentration to 1.5 m, the pore size remains barely changed, but the specific surface area and total pore volume increase to ∼357 m2 g−1 and 0.75 cm3 g−1, respectively. In general, cationic surfactants are efficient for the synthesis of mesoporous silica largely due to favorable silicate anion interaction through Coulomb forces.30 CTAB micelles assemble on the surface of hydrophobic ligand-capped nanocrystals through hydrophobic–hydrophobic interaction. When TEOS molecules are induced, negatively charged oligomeric silicate species approach the spherical micelle surface via Coulomb forces.31
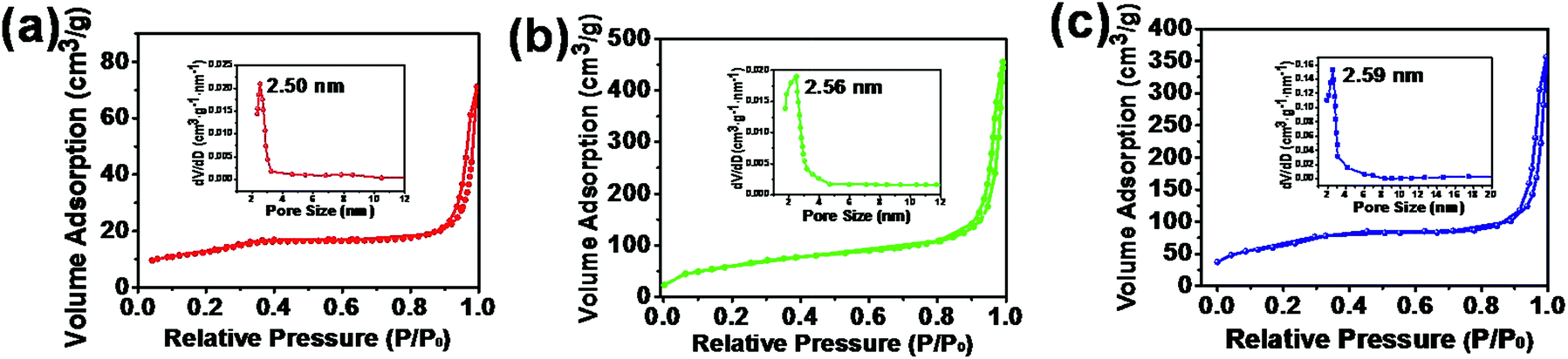 | ||
| Fig. 5 N2 adsorption–desorption isotherms and pore size distribution (the inset) of (a) STO:Yb3+,Er3+@0.5mSiO2, (b) STO:Yb3+,Er3+@1.0mSiO2 and (c) STO:Yb3+,Er3+@1.5mSiO2 nanoparticles. | ||
| Samples | SBET (m2 g−1) | VP (cm3 g−1) | DP (nm) |
|---|---|---|---|
| STO:Yb3+,Er3+@0.5mSiO2 | ∼90 | 0.25 | ∼2.50 |
| STO:Yb3+,Er3+@1.0mSiO2 | ∼222 | 0.59 | ∼2.56 |
| STO:Yb3+,Er3+@1.5mSiO2 | ∼357 | 0.75 | ∼2.59 |
The concentration of the core and TEOS are the crucial factors of the shell structure. As only the concentration of CTAB was changed during the synthesis, the size and morphology are barely changed.17,18,31 The increased CTAB concentration facilitates the absorption of TEOS, which is critical for the formation of uniform mSiO2 shell. In consequence, the specific surface area and total pore volume increases with the increased amount of CTAB. However, the pore size does not change distinguishably because of the short chain of CTAB molecules, which agrees with the previous literature.28 For the photoluminescent properties, the photoluminescent intensity decreases with the increased concentration of CTAB (Fig. S4†). This is attributed to the increased surface area and pore volume which induce more defects that may quench the spectral intensity.20
The cytotoxicity of the carriers STO:Yb3+,Er3+@mSiO2 was measured by in vitro MTT assay using L929 fibroblast. As shown in Fig. 6, more than 94% L929 fibroblast cell viability was obtained with varied particle concentration, reflecting that STO:Yb3+,Er3+@mSiO2 NPs, even at a relatively high concentrations, do not present negative effect to the cyto-compatibility.
 | ||
| Fig. 6 Cell viability of L929 fibroblast cells incubated with different concentration of STO:Yb3+,Er3+@mSiO2 nanoparticles for 48 hours. | ||
3.2. Drug loading properties
During DOX drug loading procedure, all types of NPs synthesized with the FT-IR spectra. As shown in the FT-IR spectra of STO:Yb3+,Er3+@1.0mSiO2 nanoparticles before and after DOX loading procedure (Fig. 7a), only absorption bands at 1086 cm−1 and 1720 cm−1, signing for the Si–O–Si bonding, present. After loading procedure, the peaks of C–O between 1200 cm−1 and 1600 cm−1 present, indicating the successful loading of DOX molecules. Furthermore, thermogravimetry (TG) was utilized to quantify the drug loading capacity. As shown in Fig. 7b, STO:Yb3+,Er3+@0.5mSiO2 NPs present ∼10% wt weight loss during the heating at 650 °C, reflecting its DOX loading capacity. With increased CTAB concentration, the DOX loading capacity of STO:Yb3+,Er3+@1.0mSiO2 and STO:Yb3+,Er3+@1.5mSiO2 nanoparticles increases to ∼18% wt and ∼24% wt, respectively. The positive correlation between NPs drug loading and incorporated CTAB content (during synthesis) can be attributed to proportional increase in pore volume and BET surface area.20 STO:Yb3+,Er3+@1.5mSiO2 NPs possess the highest pore volume and BET surface area, and silanol (Si–OH) groups also present in the highest quantity at the pore surface of mesoporous silica. Hence, it bonds highest concentration of DOX molecules via such increased hydrogen bonds. Furthermore, the luminescent intensity of STO:Yb3+,Er3+@mSiO2 before and after DOX loading was compared. As shown in Fig. S5,† all three types of nanoparticles present a rather similar variation trend. The photoluminescent intensity decreases in a magnificent extent after DOX loading procedure. This is induced by the quench effect of high phonon frequency organic groups of DOX molecules. | ||
| Fig. 7 (a) FTIR spectra of DOX and DOX loaded STO:Yb3+,Er3+@1.0mSiO2 particles; (b) TG curves of DOX loaded: (1) STO:Yb3+,Er3+@0.5mSiO2; (2) STO:Yb3+,Er3+@1.0mSiO2; (3) STO:Yb3+,Er3+@1.5mSiO2 nanoparticles. | ||
3.3. Drug release and its optical responses
The cumulative release of DOX from DOX–STO:Yb3+,Er3+@mSiO2 (in PBS) systems as a function of time are shown in Fig. 8. The releasing phenomena from three types of particles exhibit dramatically different releasing manner. For the STO:Yb3+,Er3+@0.5mSiO2 NPs, within the initial 5 hours, as high as ∼60% wt of the total DOX loaded was released from the nanoparticles. After 60 hours, ∼75% wt of the drug was released. In contrast, when the CTAB concentration was set at 1.5 m, only ∼40% wt of DOX loaded was released, and ∼50% wt of the DOX molecules was liberated from the NPs after 60 hours. Although STO:Yb3+,Er3+@1.5mSiO2 NPs presented the highest drug loading capacity, it still possessed the most sustained drug release rate due to its highest pore volume. The DOX release kinetics has therefore been successfully manipulated via the control of the mesoporous structure of STO:Yb3+,Er3+@mSiO2 nanoparticles. | ||
| Fig. 8 Cumulative release of DOX from three types of STO:Yb3+,Er3+@mSiO2 nanoparticles in PBS. | ||
While the controllable DOX releasing and loading have been achieved, investigating the release kinetics relationship with UC emission intensity is highly valued at a stage where theranostic therapies provide more accurate delivery and targeting of actives. The excitation (980 nm spectrum, power density 100 mW cm−2) of the particles may induce increased temperature of its surrounding microenvironment with a certain magnitude. However, such effect is unlikely to induce unexpected decomposition of DOX or ceramic particles, as confirmed by other studies.4,5,32
In this study, it was found that photoluminescent phenomenon also varied significantly during the drug release progress. As shown in Fig. 9a–c, under the continuous laser excitation at 980 nm, the 662 nm emission is remarkably enhanced for all samples during DOX molecule liberation. The optical intensity of the emission at 662 nm of all three particles is also plotted as the function of DOX release time (Fig. 9d). At three different predetermined time intervals (0 h, 40 h, 60 h), the nanoparticles were collected and air dried. The optical images were recorded directly on the sufficient sample powder under the excitation of 980 nm spectrum. The inset images confirm that the red color of the samples changes from dark to bright, which visually confirms the changing trend of UCL phenomenon during drug release.
 | ||
| Fig. 9 The UCL spectra of (a) STO:Yb3+,Er3+@0.5mSiO2; (b) STO:Yb3+,Er3+@1.0mSiO2; (c) STO:Yb3+,Er3+@1.5mSiO2 nanoparticles during drug releasing (the insets are the optical images of nanoparticles released for different time); (d) the UCL emission intensity evolution of STO:Yb3+,Er3+@mSiO2 nanoparticles during drug release. | ||
Although the emission is enhanced during the drug releasing for all particles, it does exhibit dramatically different enhancing manner. As expected, the STO:Yb3+,Er3+@0.5mSiO2 nanoparticles with fastest drug release kinetics show the rapid optical enhancement, and in contrast the STO:Yb3+,Er3+@1.5mSiO2 NPs which induce the most sustained DOX releasing phenomenon present the slowest optical enhancement.
It is known that the emission of rare earth ions can be quenched in the environment with high phonon frequency.8 The DOX molecules consist of various organic groups with high vibration frequencies from 1000 cm−1 to 3500 cm−1 (Fig. S6†), and play a vital role in quenching the photoluminescent emission. It has been documented that the DOX molecules weaken the red emission from 4F9/2 level and also the green emission from 2H11/2 and 4S3/2 levels.8,20 During the drug releasing process, DOX molecules are gradually liberated from the particles, and thus such quenching effect is weakened, resulting in the increase of emission intensity. More rapid releasing phenomenon induces faster light intensity enhancement. The direct relationship between the up-conversion emission intensity and extent of DOX release can be potentially utilized for monitoring the drug release process from a delivery system into its surrounding tissue or host environment. One notable fact is that the particles synthesized show relatively low up-conversion efficiency due to several possible reasons (not-optimal Er/Yb content, size effect et al.). The improvement of upconversion properties of the particles could facilitate its functionality of DOX delivery with optical-monitored releasing achieved in this study.
4. Conclusions
A variety of photoluminescent STO:Yb3+,Er3+@mSiO2 core–shell mesoporous nanoparticles were successfully synthesized for the anti-tumor agent (DOX) delivery for the first time. With the increased concentration of surfactant CTAB from 0.0275 M to 0.0825 M, the nanoparticles present increased surface area and pore volume, and in consequence the DOX loading capacity was doubled. The in vitro cytotoxicity assessment confirms the cyto-compatibility of STO:Yb3+,Er3+@mSiO2 nanoparticles synthesized.Meanwhile, the STO:Yb3+,Er3+@1.5mSiO2 nanoparticles which induced the highest drug loading capacity exhibited the most sustained DOX releasing kinetics. More importantly, it was found that the up-conversion luminescent intensity of the particles significantly enhanced with the progress of the DOX releasing. More rapid drug release induces more rapid photoluminescent enhancement, and vice versa. This is due to the organic groups with high phonon frequency of DOX molecules. Although several challenges exist before they can be adopted for clinical use, STO:Yb3+,Er3+@mSiO2 nanoparticles are promising materials for novel drug delivery systems possessing in situ and real-time tracking diagnosis and therapeutics.
Acknowledgements
This work was financially supported by the National Nature Science Foundation of China (No. 51232006) and the Nature Science Foundation of Zhejiang Province (No. LY15E020005).Notes and references
- L. P. Cristina, T. Julie and V. Rita, Adv. Drug Delivery Rev., 2015, 75, 81 Search PubMed.
- S. H. Nam, Y. M. Bae, Y. I. Park, J. H. Kim, H. M. Kim, J. S. Choi, K. T. Lee, T. Hyeon and Y. D. Suh, Angew. Chem., Int. Ed., 2011, 50, 6093 CrossRef CAS PubMed.
- X. Y. Ji, F. Peng, Y. L. Zhong, Y. Y. Su and Y. He, Colloids Surf., B, 2014, 124, 132–139 CrossRef CAS PubMed; D. Wu, A. B. Descalzo, F. Weik, F. Emmerling, Z. Shen, X. Z. You and K. Rurack, Angew. Chem., Int. Ed., 2008, 47, 193 CrossRef PubMed.
- J. Shen, L. Zhao and G. Han, Adv. Drug Delivery Rev., 2013, 65, 744 CrossRef CAS PubMed; X. Li, J. Zhu, Z. Man, Y. Ao and H. Chen, Sci. Rep., 2014, 4, 4446 Search PubMed.
- A. M. Smith, M. C. Mancini and S. Nie, Nat. Nanotechnol., 2009, 4, 710 CrossRef CAS PubMed; R. Weissleder and V. Ntziachristos, Nat. Med., 2003, 9, 123 CrossRef PubMed; X. Zhang, P. P. Yang, Y. L. Dai, P. A. Ma, X. J. Li, Z. Y. Cheng and J. Lin, Adv. Funct. Mater., 2013, 23, 4067 CrossRef.
- X. Wang, X. G. Kong, Y. Yu, Y. J. Sun and H. Zhang, J. Phys. Chem. C, 2007, 111, 15119 Search PubMed; J. C. G. Bunzli, Chem. Rev., 2010, 110, 2729 CrossRef CAS PubMed.
- J. Zhou, Z. Liu and F. Y. Li, Chem. Soc. Rev., 2012, 41, 1323 RSC.
- Z. Y. Hou, C. X. Li, P. A. Ma, G. G. Li, Z. Y. Cheng, C. Peng and J. Lin, Adv. Funct. Mater., 2011, 21, 2356 CrossRef CAS.
- S. Jiang and Y. Zhang, Langmuir, 2010, 26, 6689 CrossRef CAS PubMed.
- N. Lewinski, V. Colvin and R. Drezek, Small, 2008, 4, 26 CrossRef CAS PubMed; S. Sharifi, S. Behzadi, S. Laurent, M. L. Forrest, P. Stroevee and M. Mahmoudi, Chem. Soc. Rev., 2012, 41, 2323 RSC.
- J. Braux, F. Velard, C. Guillaume, S. Bouthors, E. Jallot, J. M. Nedelec, D. L. Maquin and P. Laquerriere, Acta Biomater., 2011, 7, 2593 CrossRef CAS PubMed; M. Schumacher, A. Lode, A. Helth and M. Gelinsky, Acta Biomater., 2013, 9, 9547 CrossRef PubMed.
- Y. C. Xin, J. Jiang, K. F. Huo, T. Hu and P. K. Chu, ACS Nano, 2009, 3, 3228 CrossRef CAS PubMed.
- L. L. Dong, Q. Luo, K. Cheng, H. Shi, Q. Wang, W. J. Weng and W. Q. Han, Sci. Rep., 2014, 4, 5084 CAS.
- J. W. Shi, J. H. Ye, L. J. Ma, S. X. Ouyang, D. W. Jing and L. J. Guo, Chem.–Eur. J., 2012, 18, 7543 CrossRef CAS PubMed.
- X. Wang, J. Zhuang, Q. Peng and Y. D. Li, Nature, 2005, 437, 121 CrossRef CAS PubMed.
- Y. H. Deng, D. W. Qi, C. H. Deng, X. M. Zhang and D. Y. Zhao, J. Am. Chem. Soc., 2008, 130, 28 CrossRef CAS PubMed.
- J. Kim, H. S. Kim, N. Lee, T. Kim, H. Kim, T. Yu and T. Hyeon, Angew. Chem., Int. Ed., 2008, 47, 8438 CrossRef CAS PubMed.
- C. T. Wu, W. Fan and J. Chang, J. Mater. Chem. B, 2013, 1, 2710 RSC.
- X. J. Kang, Z. Y. Cheng, C. X. Li, D. M. Yang, M. M. Shang, P. A. Ma and J. Lin, J. Phys. Chem. C, 2011, 115, 15801 CAS.
- F. C. D. Lemos, J. E. C. da Silva, D. M. A. Melo, M. S. C. Câmara, P. S. de Lima and C. E. J. Carneiro, Inorg. Mater., 2008, 44, 866 CrossRef CAS.
- R. Pazik, M. Maczka, M. Malecka, L. Marciniak, A. Ekner-Grzyb, L. Mrowczynska and R. J. Wiglusz, Dalton Trans., 2015, 44, 10267 RSC.
- Y. Kinemuchi, K. I. Mimura, A. Towata and K. Kato, J. Electron. Mater., 2014, 43, 2011 CrossRef CAS.
- S. V. Eliseeva and J. C. G. Bünzli, Chem. Soc. Rev., 2010, 39, 189 RSC.
- K. Binnemans, Chem. Rev., 2009, 109, 4283 CrossRef CAS PubMed.
- Z. Y. Hou, C. X. Li, P. A. Ma, Z. Y. Cheng, X. J. Li, X. Zhang, Y. L. Dai, D. M. Yang, H. Z. Lian and J. Lin, Adv. Funct. Mater., 2012, 22, 2713 CrossRef CAS.
- J. F. Liu and Y. D. Li, Adv. Mater., 2007, 19, 1118 CrossRef CAS.
- H. Y. Fan, K. Yang, D. M. Boye, T. Sigmon, K. J. Malloy, H. F. Xu, G. P. López and C. J. Brinker, Science, 2004, 304, 567 CrossRef CAS PubMed; H. Y. Fan, W. L. Erik, S. Chessa, G. John, T. David, B. Scott, B. Tim, C. W. Michael and C. J. Brinker, Nano Lett., 2005, 5, 645 CrossRef PubMed; L. Zhang, S. Z. Qiao, Y. G. Jin, H. G. Yang, S. Budihartono, F. Stahr, Z. F. Yan, X. L. Wang, Z. P. Hao and G. Q. Lu, Adv. Funct. Mater., 2008, 18, 3203 CrossRef.
- A. M. Vaz, D. Serrano-Ruiz, M. Laurenti, P. Alonso-Cristobal, E. Lopez-Cabarcos and J. Rubio-Retama, Colloids Surf., B, 2014, 114, 11 CrossRef CAS PubMed.
- Y. F. Zhu, E. Kockrick, T. Ikoma, N. Hanagata and S. Kaskel, Chem. Mater., 2009, 21, 2547 CrossRef CAS.
- D. Y. Zhao, J. L. Feng, Q. S. Huo, N. Melosh, G. H. Fredrickson, B. F. Chmelka and G. D. Stucky, Science, 1988, 279, 548 CrossRef.
- W. Li, Q. Yue, Y. H. Deng and D. Y. Zhao, Adv. Mater., 2013, 25, 5129 CrossRef CAS PubMed.
- J. N. Liu, W. B. Bu, L. M. Pan, S. J. Zhang, F. Chen, L. P. Zhou, K. L. Zhao, W. J. Peng and J. L. Shi, Biomaterials, 2012, 33, 7282 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra27459a |
| This journal is © The Royal Society of Chemistry 2016 |