DOI:
10.1039/C5RA27426E
(Paper)
RSC Adv., 2016,
6, 13928-13933
Copper doped EuMnO3: synthesis, structure and magnetic properties†
Received
22nd December 2015
, Accepted 11th January 2016
First published on 14th January 2016
Abstract
A solid solution EuMn1−xCuxO3−δ (0 ≤ x ≤ 0.316) was synthesized by a traditional solid state reaction at 1150 °C. It crystallized into the Pnma space group, which was confirmed by powder X-ray and selected area electron diffraction. Magnetic susceptibility measurements showed that the two magnetic transitions for these materials occurred from 52 to 22 K and 45 to 20 K.
Introduction
Many studies have been performed on LnMnO3 (Ln = La, Pr, Nd, Sm, Eu, Gd, Tb, Dy, Ho, Er, Tm, Yb, and Lu) for their interesting possible applications such as LaxSr1−xMnO3−δ (ref. 1) studied for cathodes of solid oxide fuel cells, Ln1−xAxMnO3−δ (A = Ca, Sr, Ba, and Pb)2 for colossal magneto-resistances (CMR), and TbMnO3 (ref. 3) for multiferroic behavior. EuMnO3 is similar to TbMnO3, and has also been studied as a multiferroic.4 At room temperature, EuMnO3 was reported to crystallize in the Pnma space group (a = 5.842, b = 7.451, and c = 5.336 Å), and was paraelectric (PE) and paramagnetic (PM).5–7 Using a single crystal sample,8 very obvious peaks were found at around 51 K and 46 K in the C/T (specific heat divided by temperature) vs. T curve and the M (magnetization) vs. T curve of EuMnO3, which was attributed to a phase transition from PM to ICAFM (incommensurate antiferromagnetic) at ∼50 K and a further transition from ICAFM to cAAFM (canted A-type antiferromagnetic) ordering at ∼44 K.9,10 When a powder sample was used, the corresponding peaks in the C/T vs. T curve were found to be around 50 K and 43 K by W. S. Ferreira et al.,11 or around 47 K and 35 K by Y. M. Mukovskii et al.6 The FC (field cooling) M vs. T curve of powder EuMnO3 provided by W. S. Ferreira et al.11 showed changes around 55 K and 40 K. The data reported by I. O. Troyanchuk et al.12 showed that the peak temperature increased with decreased oxygen content. These disagreements may be easily understood when one accepts the results reported by O. M. Fedorova et al.13 that there is a solid solution Eu1−xMn1+xO3. Similar studies were also reported for La1−xMnO3,14 Pr1−xMn1+xO3,15 and Tb1−xMnyMnO3.16,17 In this case, it is reasonable to accept that the real composition of the sample used by different researchers was different, which resulted in different observations.
It is well known that A- and B-site doping could strongly change the properties of the rare earth manganites LnMnO3.1,2,18–22 Since then, several elements, such as Ca, Sr, Y, La, Tb, and Lu, have been tried to substitute Eu in EuMnO3.4,6,12,23–29 The published data shows that when the radius of the doping ions (such as Ca2+, Sr2+, and La3+) was larger than that of Eu3+, the peak temperature of the ZFC curve of M–T for Eu1−xAxMnO3 (A = Ca, Sr, and La) shifted to high temperature with the increased doping element;6,11,23–25 and when the radius of the doping ions (such as Y3+, Tb3+, and Lu3+) was smaller than that of Eu3+, the corresponding transition temperatures shifted to low temperature with the increased doping element content.4,26–29 It is interesting that to date only I. O. Troyanchuk et al.30–32 have reported the synthesis and properties of EuMn1−xMxO3 (M = Cr, Co, and Ni), which showed that when x < 0.2 the magnetic phase transition occurred at lower temperature than that for EuMnO3, and when x > 0.2 the magnetic phase transition occurred at higher temperature. To broaden the knowledge of EuMn1−xMxO3, the synthesis and characterization of EuMn1−xMxO3 with M other than Cr, Co, and Ni, was tried by us. Herein, we will report the structure and magnetic properties of EuMn1−xCuxO3.
Experimental
The samples with the nominal formula EuMn1−xCuxO3−δ (x = 0.00, 0.10, 0.20, 0.30, 0.35, and 0.50, named as C1, C2, C3, C4, C5, and C6, respectively) were synthesized from stoichiometric amounts of Eu2O3 (99.95%), CuO (A.R.), and MnCO3 (A.R.). The oven-dried reagents were mixed and homogenized by grinding for about thirty minutes with an agate mortar and a pestle. The mixtures were subjected to 6 h calcinations at 800 °C and 900 °C. They were then pressed into pellets to undergo four 12 h heat treatments at 1150 °C followed by a furnace cooling every time with intermediate grinding. All the treatments were done in air. The weights of the samples were monitored before and after heat treatments. The maximum difference was about 4 mg for the 6 g samples. Therefore, the compositions of the samples were considered to be the same as the initial ones.
Powder X-ray diffraction (PXRD) data were collected on a PANalytical X'Pert3 Powder with Cu Kαα (λ1 = 0.15405 nm and λ2 = 0.15443 nm) radiation (2θ range: 5–120° for 10 hours; step: 0.0131) at 40 kV and 40 mA at room temperature. The X-ray diffraction data were analyzed using GSAS33,34 software. The magnetic properties were investigated with a cryogenic physical property measurement system (PPMS) from 2 to 300 K. Selected area electron diffractions (SAED) were carried out on JEM2100F with a 200 kV accelerating voltage. The X-ray photoelectron spectroscopy (XPS) patterns were obtained with a UK Kratos Axis Ultra spectrometer with an Al Kα X-ray source operated at 15 kV and 15 mA. The chamber pressure was less than 5.0 × 10−9 Torr. Electron binding energies were calibrated against the C 1s emission at Eb = 284.8 eV.
Results and discussion
Structure and solid solution of EuMn1−xCuxO3−δ
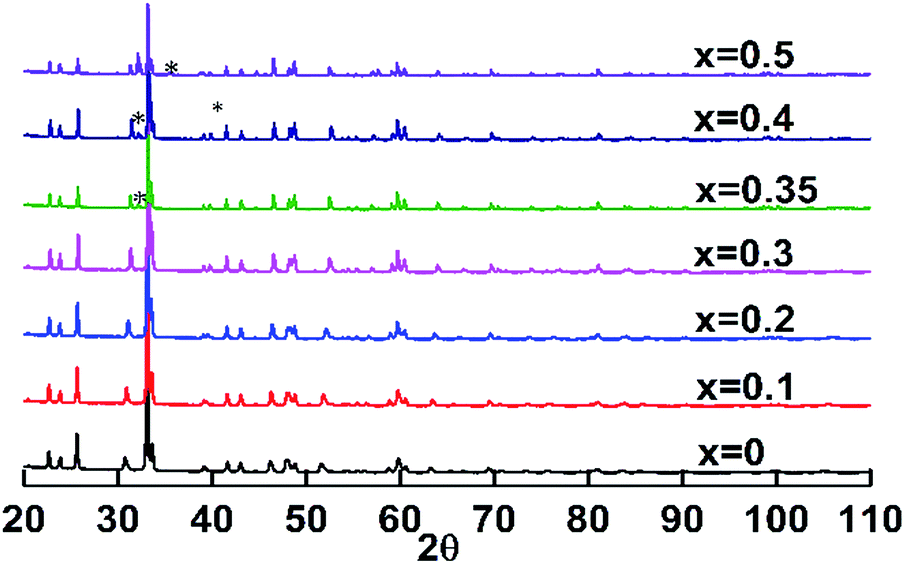
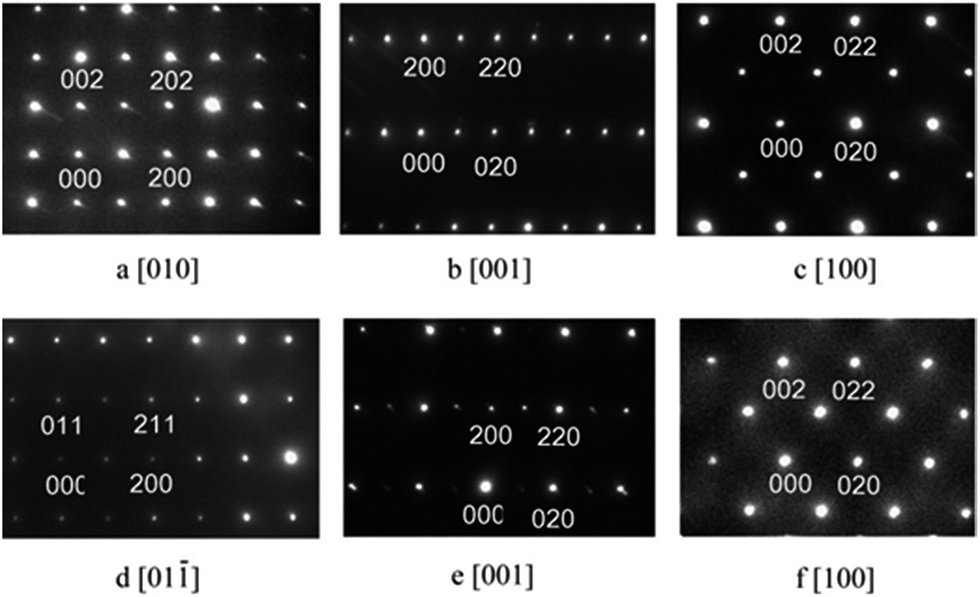
The X-ray diffraction patterns of C1 to C4 were very similar to that reported for EuMnO3,5–7 as shown in Fig. 1. For C5 or C6, reflections of the impurity could be found around about 32°. Selected area electron diffraction patterns of C1 to C4 confirmed that they all crystallized in the Pnma space group. Typical patterns are shown in Fig. 2.
 |
| | Fig. 1 Powder X-ray diffraction patterns of the samples with the nominal formula EuMn1−xCuxO3−δ (0 ≤ x ≤ 0.50). *, reflections for the impurity. | |
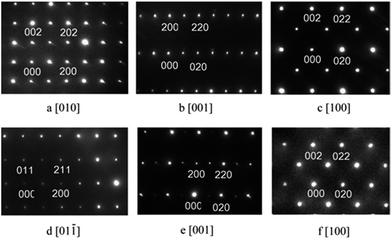 |
| | Fig. 2 Selected area electron diffraction patterns of C1 (a–c) and C4 (d–f). | |
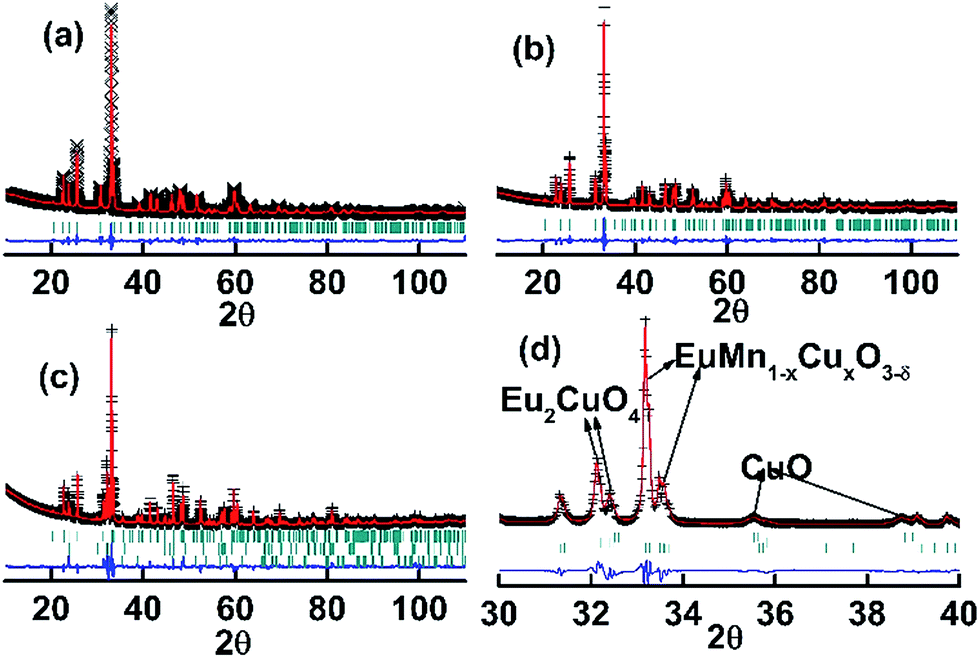
Rietveld refinements were performed for the X-ray diffraction data of C1 to C6. For the samples with x > 0, Cu was supposed to randomly sit at the site of Mn. Good fits were obtained (the corresponding data listed in Table S1 in ESI†) and typical Reitveld plots are shown in Fig. 3. For C5 and C6, a small amount of Eu2CuO4 (ref. 35) and CuO36 was found, which is marked in Fig. 3d. Then the three phases model was used to refine the data of C5 and C6.
 |
| | Fig. 3 Rietveld plots of powder X-ray diffraction patterns for the sample C1 (a), C4 (b) and C6 (c and d). The symbol + represents the observed value, the solid line represents the calculated value, the marks below the diffraction patterns are the calculated reflection positions, and the difference curve is shown at the bottom of the figure. | |
Increasing Cu in EuMn1−xCuxO3−δ led to a decrease in the a-axis and b-axis and an increase in the c-axis, as shown in Fig. 4, which agreed well with Vegard's law, which is as follows:37,38
| | |
aOx = aO0(1 − x) + aO1x
| (1) |
| | |
bOx = bO0(1 − x) + bO1x
| (2) |
| | |
cOx = cO0(1 − x) + cO1x
| (3) |
where
aOx (
bOx,
cOx),
aO0 (
bO0,
cO0),
aO1 (
bO1,
cO1) are the lattice parameters
a (
b,
c) of the orthorhombic EuMn
1−xCu
xO
3−δ, pure orthorhombic EuMnO
3 and supposed orthorhombic EuCuO
3. ‘
x’ were the composition variable as given by Cu/(Cu + Mn). According to the data shown in
Fig. 4, the maximum
x may be 0.316. Then the solid solution for our samples was EuMn
1−xCu
xO
3−δ with 0 ≤
x ≤ 0.316. The lattice parameters of the solid solution EuMn
1−xCu
xO
3−δ follow the relationship of
b/√2<
c <
a, which means the structure belongs to orthorhombic OI as mentioned by several researchers.
5
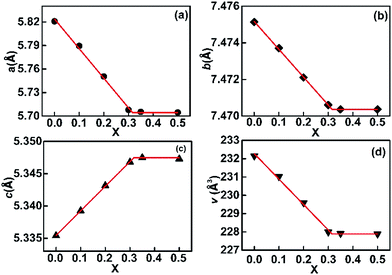 |
| | Fig. 4 Variation of the lattice parameters a, b and c and the volume of the unit cell V of the solid solution EuMn1−xCuxO3−δ in the samples. X = Cu/(Cu + Mn) in the samples. | |
XPS data for EuMn1−xCuxO3−δ
XPS spectra of C1, C2, C3, and C4 have been obtained to estimate the valence state of Eu, Mn and Cu in the solid solution EuMn1−xCuxO3−δ. The XPS spectra for the europium in the four samples were almost the same, as shown in Fig. 5a. The two peaks are the signals of Eu 3d5/2 and Eu 3d3/2 with binding energies at 1135.6 eV and 1165.2 eV, respectively, which indicates that the valence state of Eu was +3.39,40 The shape of the spectrum for the Cu 2p level was similar for C2 to C4 with the shake-up satellite peak. This indicates that the valence state of Cu was +2.40 The peak position of the main peak was at 933.7 eV, which is very close to that for Cu in CuO41 and LaMn1−xCuxO3−δ.42 As reported, the binding energies of peak positions of the Mn 2p3/2 level of α-Mn2O3 and β-MnO2 were very close, 641.9 and 642.2 eV,43 the peak position of the Mn 2p3/2 level of MnO was 640.6 eV with satellite peaks.43 Therefore, the spectra for the Mn 2p level of EuMn1−xCuxO3−δ at x = 0, 0.1, 0.2, and 0.3 shown in Fig. 5 could be treated as the combination of Mn3+ and Mn4+ (the corresponding binding energies listed in Table S3 in the ESI†). The obtained ratio of Mn4+![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Mn3+ by analyzing the XPS spectra agreed well with the expected ratio of Mn4+:Mn3+ when supposing that each Cu2+ needs a Mn4+ to balance the charge of the compounds.
:Mn3+ by analyzing the XPS spectra agreed well with the expected ratio of Mn4+:Mn3+ when supposing that each Cu2+ needs a Mn4+ to balance the charge of the compounds.
 |
| | Fig. 5 XPS spectra of Eu (a), Cu (b) and Mn (c–f) in the samples. | |
Magnetic property
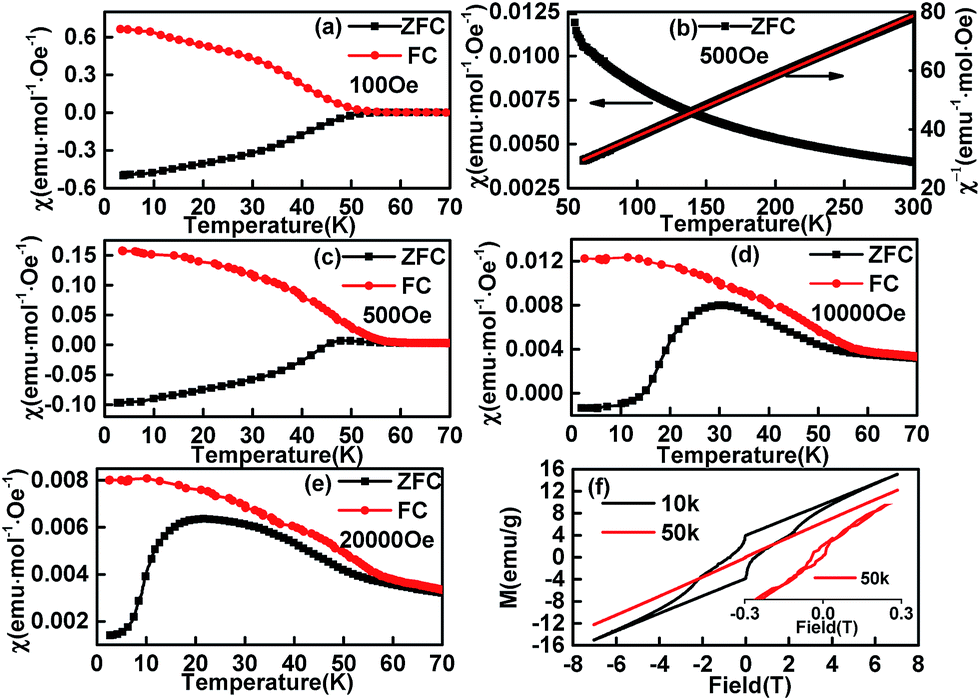
The temperature dependence of magnetization of the sample C1 in ZFC (zero field cooling) and FC modes are shown in Fig. 6. The FC curve departures from the ZFC curve at ∼52 K, which is attributed to the PM-to-ICAFM9 or PM-to-AFM6 transition, and is noted as TN1. It was hard to get the neutron diffraction data for EuMnO3 (ref. 44 and 45) because the neutron can be absorbed by Eu and the magnetic structure of EuMnO3 has not been solved to date, i.e. at present it is difficult to know if the magnetic phase for EuMnO3 around 52 K was ICAFM or AFM, although T. Goto et al.9 predicted it should be ICAFM. Therefore, it is better to note it as AFM because ICAFM is one type of AFM.
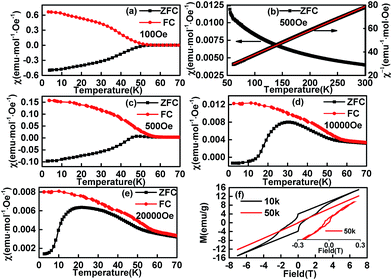 |
| | Fig. 6 Temperature dependent magnetization of C1 measured at several field ((a) 100 Oe, (b) 500 Oe, (c) 500 Oe, (d) 10000 Oe, (e) 20000 Oe) and field dependent magnetization of C1 at different temperature (f). | |
Above TN1, as shown in Fig. 6c, the temperature dependent invert magnetic susceptibility can be fitted to a straight line, which reveals that the compound followed Curie–Weiss behaviour (χ = C/(T − Tθ), C = 4.87 for C1). The obtained effective magnetic moment (μeff =  ) was 6.24 μB, which is similar to Y. Tadokoro's 5.9 μB24 Y. Chen's 6.14 μB,46 A. V. Gavrikov's 6.17 μB (ref. 47) and T. S. Chan's 6.61 μB.29 The manganese contribution to EuMnO3 could be μ(Mn3+) = 4.9 μB, which are more and less than 6.24 μB. Therefore part of the magnetization must be from Eu3+. It is known that for a VanVleck Eu3+ ion (7Fo; μ = 0) an admixture of the J = 0 and J = 1 levels may lead to a non-negligible magnetic moment of 3.4–3.6 μB.48 Then the total effective magnetic moment μeff for C1 (EuMnO3) could be 5.96–6.08 μB (μeff2 = μ2(Mn3+) + μ2(Eu3+)), and agreed well with the experimental value.
) was 6.24 μB, which is similar to Y. Tadokoro's 5.9 μB24 Y. Chen's 6.14 μB,46 A. V. Gavrikov's 6.17 μB (ref. 47) and T. S. Chan's 6.61 μB.29 The manganese contribution to EuMnO3 could be μ(Mn3+) = 4.9 μB, which are more and less than 6.24 μB. Therefore part of the magnetization must be from Eu3+. It is known that for a VanVleck Eu3+ ion (7Fo; μ = 0) an admixture of the J = 0 and J = 1 levels may lead to a non-negligible magnetic moment of 3.4–3.6 μB.48 Then the total effective magnetic moment μeff for C1 (EuMnO3) could be 5.96–6.08 μB (μeff2 = μ2(Mn3+) + μ2(Eu3+)), and agreed well with the experimental value.
Below TN1, when the applied and measured field was about 100 Oe, the temperature dependent ZFC and FC curves were almost symmetric with a mirror-reflection. When the applied field was about 500 Oe, a hump shaped anomaly appeared around 45 K, which may be related to the second magnetic phase transition AFM-to-cAFM (herein, cAFM means canted antiferromagnetic) reported for EuMnO3 and can be noted as TN2. Although it is believed that the second magnetic phase for EuMnO3 is canted A-type antiferromagnetic, the fact that no magnetic structure has been presented for EuMnO3 indicates the A-type is just a supposed suggestion instead of a true assignment. Therefore it is better to use cAFM to describe the second magnetic phase of EuMnO3. With the increased applied field (Fig. 6a, c, d and e), the peaks became more apparent and moved to lower temperatures. This was similar to that reported by Y. M. Mukovskii6 except for the negative magnetization. Negative values were observed in the ZFC curves until the measured field was up to 20000 Oe, which suggests that the zero field in the cooling case was a small negative field.49–51 A detailed discussion is presented below.
It must be stated that the present phenomenon was not a true negative magnetization. The negative magnetizations were mostly observed in the FC curves of the corresponding compounds or mixtures,52 which may be interpreted by the mean field theory (MFT) given by L. Néel53 and extended by the K. P. Belov.54 There were few examples to show negative magnetization only in ZFC case. The negative magnetization in ZFC only had been reported for UCu2Ge2,55 which was attributed to the effect of a trapped negative field in a solenoid of a superconducting magnet as we believed for our sample C1. Another example was reported by M. Mączka et al.56 for [(CH3)2ND2][M(HCOO)3] (M = Ni, Mn) without detailed discussions on such a phenomenon. The third example is Nd0.93MnO2.96,57 which had been reported to show a similar features above 20 K, wherein the Nd0.93MnO2.96 was believed to be a mixture of NdMnO3 and Nd0.9MnO3 with different magnetic ordering temperatures except for a small amount of Mn3O4. In addition, the magnetic moments of NdMnO3 and Nd0.9MnO3 were suggested to be anti-parallel coupled. Then, the negative magnetization for Nd0.93MnO2.96 in the ZFC case seems to be explained as follows: when the sample cooled down under zero field (typical a very small positive field), the first phase ordered at about 80 K with the magnetic moment paralleling to the direction of the measured field. Subsequently, when the temperature lowers down further, the second phase ordered at about 70 K with the magnetic moment anti-paralleling to the direction of the measured field. If the magnetic moment of the second phase was larger than that of the first phase at low temperature, the negative magnetization would be observed when a positive measured field is applied (typical measurement is started from low temperature). In this case, one should also observe the negative magnetization under FC condition with a little larger positive applied field. However, the experimental result was positive for Nd0.93MnO2.96 at FC case above 20 K. This may mean that the abovementioned explanation did not fit the experiment results. The abovementioned experiment results can be explained well with that the “negative magnetization” observed only at ZFC case should be caused by a small negative field served as the zero field in the zero field cooling case. This is simple and puzzle, but it is the fact. Of cause, if one wants to observe this phenomenon, the good candidate may be the material, which has strong magnetic hysteresis loops at low temperature, wherein the magnetic moment is not easily inversed by a small field. A strong loop was observed in the isothermal magnetization curve at 10 K for C1 (Fig. 6f). It was found that a high field about 20000 Oe was needed to inverse the magnetic moment at this temperature, which could explain why negative values can be observed in the ZFC curves until the measured field was up to 20000 Oe when cooled under a small negative field. The loop becomes smaller and smaller with the increase of the temperature. Up to 50 K, small loop is still observed.
When doped with Cu, the “negative magnetization” disappears, as shown in Fig. 7. Similar to C1, the temperature wherein ZFC and FC curves started to depart is attributed by us to the phase transition temperature from PM to AFM, which is noted as TN1, and the peak temperature in the ZFC curves is attributed to the phase transition temperature from AFM to cAFM, which is noted as TN2. The corresponding TN1 and TN2 was 33 K and 29 K for C2, 28 K and 23 K for C3, and 22 K and 20 K for C4. The decreased transition temperature with increased Cu in the samples could be attributed to double exchange and super exchange interactions between neighboring Mn3+(Mn4+) and Mn3+(Mn4+) ions through the path Mn3+(Mn4+)–O–Mn3+(Mn4+)51,58 was weakened because more Mn was replaced by Cu. A similar feature was also reported for EuMn1−xMxO3 (M = Cr, Co and Ni).30–32 Field dependent magnetizations of C2 to C4 are also shown in Fig. 7d–f. Compared to C1, the magnetization at 10 K became weaker when more Cu was doped, which indicated that the doped Cu weakened the magnetic interaction of the corresponding compounds. Although a small remnant magnetization was observed at T = 50 K for C1, the field dependent magnetization for other samples at 50 K was linear, which revealed a paramagnetic behavior at 50 K for C2 to C4.
 |
| | Fig. 7 Temperature dependence of the magnetization of EuMn1−xCuxO3−δ (0 ≤ x ≤ 0.3). | |
The temperature dependent inverse magnetizations (1/χ ∼ T) for C2 to C4 are shown in Fig. 7g–i above 50 K. They follow the Curie–Weiss behaviour (χ = C/(T − Tθ)). The corresponding results are summarized in Table S4 in the ESI.† Obviously, the effective moments (μeff) decreased with increasing x, which was simply due to replacement of Mn3+ (4.9 μB) ions by Cu2+ (1.73 μB) and Mn4+ (3.87 μB) ions with μeff2 = (1 − 2x) × μ2(Mn3+) + x × μ2(Mn4+) + x × μ2(Cu2+) + μ2(Eu3+).
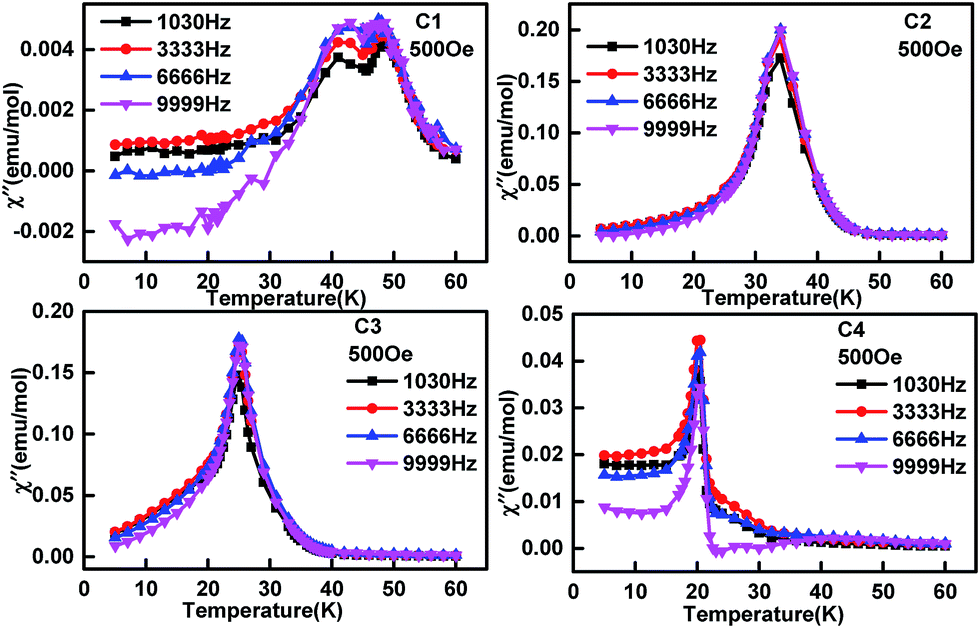
To check if there is magnetic frustration in the solid solution EuMn1−xCuxO3−δ, AC susceptibility analysis was performed for C1 to C4 with the corresponding data shown in Fig. 8 and 9. The real part of the AC magnetization (Fig. 8C1) curve of EuMnO3 showed two peaks at 47.5 and 42.3 K. The former may correspond to a PM-to-AFM6 transition. The 47.5 K temperature is little different from 52 K observed from the ZFC and FC M–T curves shown in Fig. 6. The reason may be related to differences in the measurement methods. The anomaly at 42 K is attributed to the AFM-to-cAFM transition. No peak around 24.7 K could be observed, although such a peak had been reported by R. Das et al.10 using a nano sample of EuMnO3 and it was related to the spin reorientation transition of the magnetic moment of EuMnO3.58 The imaginary part of the AC magnetization (Fig. 9C1) shows also anomalies at around 47.5 and 42.4 K. No frequency dependency was observed, which indicates that no magnetic frustration occurred in C1. When Cu was doped into the site of Mn in EuMnO3 to form the solid solution EuMn1−xCuxO3, one broad peak could be observed at AC susceptibility (Fig. 8C2–C4 and 9C2–C4) with the peak temperature of about 32 K for C2, 26 K for C3, and 20 K for C4, which is very close to the two temperatures TN1 and TN2 observed from the ZFC and FC M–T curves shown in Fig. 7. If the resolution of the AC curve could be improved, maybe two peaks could be observed, which correspond to TN1 and TN2. Therefore, these peak temperatures are related to the magnetic ordering. No frequency dependent behaviour was observed, which indicated that the magnetic frustration for C2 to C4 was too weak to be observed.
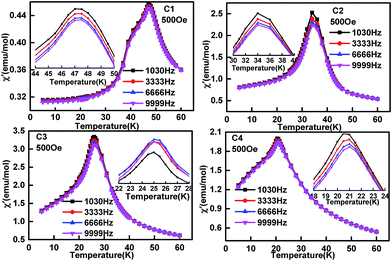 |
| | Fig. 8 Temperature dependence of real part of AC magnetization of samples EuMn1−xCuxO3−δ (0 ≤ x ≤ 0.3) with different frequencies. | |
 |
| | Fig. 9 Temperature dependence of the imaginary part of AC magnetization of samples EuMn1−xCuxO3−δ (0 ≤ x ≤ 0.3) with different frequencies. | |
Conclusions
A solid solution EuMn1−xCuxO3−δ was synthesized by a traditional solid state method. Under the present synthesis conditions, the solid solution range was 0 ≤ x ≤ 0.316. The space group for this solid solution around room temperature was suggested to be Pnma. With the increase of Cu in the samples, the temperature corresponding to the magnetic phase transition decreased in the whole solid solution, which was different from that observed in EuMn1−xMxO3 with M being other than Cr, Co, and Ni.
Acknowledgements
This study is supported by a National Key Basic Research Project of China (2010CB833103), the National Natural Science Foundation of China (Grant 21271014).
Notes and references
- N. Q. Minh, J. Am. Ceram. Soc., 1993, 76, 563 CrossRef CAS.
- A. M. Haghiri-Gosnet and J. P. Renard, J. Phys. D: Appl. Phys., 2003, 36, R127 CrossRef CAS.
- N. Aliouane, D. N. Argyriou and J. Strempfer, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 020102 CrossRef.
- J. Hemberger, F. Schrettle, A. Pimenov, P. Lunkenheimer, V. Y. Ivanov, A. A. Mukhin, A. M. Balbashov and A. Loidl, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 035118 CrossRef.
- N. V. Kasper and I. O. Troyanchuk, J. Phys. Chem. Solids, 1996, 57, 1601 CrossRef CAS.
- Y. M. Mukovskii, G. Hilscher, H. Michor and A. M. Ionov, J. Appl. Phys., 1998, 83, 7163 CrossRef CAS.
- T. Mori, N. Kamegashira, K. Shishido, T. Fukuda and T. Aoki, Mater. Lett., 2002, 54, 238 CrossRef CAS.
- T. Kimura, S. Ishihara, H. Shintani, T. Arima, K. T. Takahashi, K. Ishizaka and Y. Tokura, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 68, 060403 CrossRef.
- T. Goto, T. Kimura, G. Lawes, A. P. Ramirez and Y. Tokura, Phys. Rev. Lett., 2004, 92, 257201 CrossRef CAS PubMed.
- R. Das and P. Poddar, RSC Adv., 2014, 4, 10614 RSC.
- W. S. Ferreira, J. A. Moreira, A. Almeida, M. R. Chaves, J. P. Araújo, J. B. Oliveira, J. M. M. Da Silva, M. A. Sá, T. M. Mendonça, P. S. Carvalho, J. Kreisel, J. L. Ribeiro, L. G. Vieira, P. B. Tavares and S. Mendonça, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 054303 CrossRef.
- I. O. Troyanchuk, N. V. Samsonenko, N. V. Kasper, H. Szymczak and A. Nabialek, Phys. Status Solidi, 1997, 160, 195 CrossRef CAS.
- O. M. Fedorova, V. F. Balakirev and Y. V. Golikov, Inorg. Mater., 2007, 43, 994 CrossRef CAS.
- A. N. Ulyanov, N. E. Pismenova, D. S. Yang, V. N. Krivoruchko and G. G. Levchenko, J. Alloys Compd., 2013, 550, 124 CrossRef CAS.
- E. Pollert and Z. Jirak, J. Solid State Chem., 1980, 35, 262 CrossRef CAS.
- R. Wang, C. X. Yang, M. Fan, M. M. Wu, C. H. Wang, X. H. Yu, J. L. Zhu, J. R. Zhang, G. B. Li, Q. Z. Huang, D. F. Chen, T. N. Jin, T. Kamiyama, F. H. Liao and J. H. Lin, J. Alloys Compd., 2013, 554, 385 CrossRef CAS.
- H. Zhang, R. Flacau, J. L. Sun, G. B. Li, F. H. Liao and J. H. Lin, Inorg. Chem., 2014, 53, 4535 CrossRef CAS PubMed.
- C. Y. Zhang, T. S. Zhang, L. Ge, S. Wang and H. M. Yuan, RSC Adv., 2014, 4, 50969 RSC.
- B. Raneesh, K. Nandakumar and A. Saha, RSC Adv., 2015, 5, 12480 RSC.
- N. Zaidi, S. Mnefgui, J. Dhahria and E. K. Hlilb, RSC Adv., 2015, 5, 31901 RSC.
- B. Singh, RSC Adv., 2015, 5, 39938 RSC.
- A. M. Farid, H. Zhang, X. H. Lin, A. M. Yang, S. H. Yang, G. B. Li, F. H. Liao and J. H. Lin, RSC Adv., 2015, 5, 12866 RSC.
- I. O. Troyanchuk, N. V. Samsonenko, H. Szymczak and A. Nabialek, J. Solid State Chem., 1997, 131, 144 CrossRef CAS.
- Y. Tadokoro, Y. J. Shan, T. Nakamura and S. Nakamura, Solid State Ionics, 1998, 108, 261 CrossRef CAS.
- H. C. Ku, C. T. Chen and B. N. Lin, J. Magn. Magn. Mater., 2004, 272, 85 CrossRef.
- J. Oliveira, J. A. Moreira, A. Almeida, M. R. Chaves, J. M. M. da Silva, J. B. Oliveira, M. A. Sa, P. B. Tavares, R. Ranjith and W. Prellier, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 094414 CrossRef.
- K. F. Wang, Q. M. Liu, S. J. Luo, Y. Wang and J. M. Liu, Thin Solid Films, 2010, 518, E12 CrossRef.
- D. A. Mota, Y. R. Barcelay, P. B. Tavares, M. R. Chaves, A. Almeida, J. Oliveira, W. S. Ferreira and J. A. Moreira, J. Phys.: Condens. Matter, 2013, 25, 235602 CrossRef CAS PubMed.
- T. S. Chan, R. S. Liu, C. C. Yang, W. H. Li, Y. H. Lien, C. Y. Huang and J. F. Lee, J. Phys. Chem. B, 2007, 111, 2262 CrossRef CAS PubMed.
- I. O. Troyanchuk, H. Szymczak, N. V. Samsonenko and A. Nabialek, Phys. Status Solidi A, 1996, 157, 167 CrossRef CAS.
- I. O. Troyanchuk, H. Szymczak, N. V. Samsonenko, A. Nabialek and E. F. Shapovalova, Phys. Status Solidi A, 1997, 163, 215 CrossRef CAS.
- I. O. Troyanchuk, N. V. Samsonenko, N. V. Kasper, H. Szymczak and A. Nabialek, Phys. Status Solidi A, 1997, 160, 195 CrossRef CAS.
- H. M. Rietveld, J. Appl. Crystallogr., 1969, 2, 65 CrossRef CAS.
- A. C. Larson and R. B. von Dreele, Report LAUR 86-748, Los Alamos National Laboratory, Los Alamos, NM, 1985 Search PubMed.
- P. Vigoureux, A. Gukasov, S. N. Barilo and D. Zhigunov, Phys. B, 1997, 234, 815 CrossRef.
- M. Ain, A. Menelle, B. M. Wanklyn and E. F. Bertaut, J. Phys.: Condens. Matter, 1992, 4, 5327 CrossRef CAS.
- L. Vegard, Z. Phys., 1921, 5, 17 CrossRef CAS.
- L. Vegard, Z. Kristallogr., 1928, 67, 239 Search PubMed.
- Y. Uwamino, Y. Ishizuka and H. Yamatera, J. Electron Spectrosc. Relat. Phenom., 1984, 34, 69 CrossRef.
- D. F. Mullica, C. K. C. Lok, H. O. Perkins, G. A. Benesh and V. J. Young, J. Electron Spectrosc. Relat. Phenom., 1995, 71, 1 CrossRef CAS.
- J. Ghijsen, L. H. Tjeng, J. van Elp, H. Eskes, J. Westerink, G. A. Sawatzky and M. T. Czyzyk, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 38, 11322 CrossRef CAS.
- K. Tabata, Y. Hirano and E. Suzuki, Appl. Catal., A, 1998, 170, 245 CrossRef CAS.
- M. Oku, K. Hirokawa and S. Ikeda, J. Electron Spectrosc. Relat. Phenom., 1975, 7, 465 CrossRef CAS.
- V. Katari, S. Achary and S. Deshpande, J. Phys. Chem. C, 2014, 118, 17900 CAS.
- B. Dabrowski, S. Kolesnik, A. Baszczuk, O. Chmaissem, T. Maxwell and J. Mais, J. Solid State Chem., 2005, 178, 629 CrossRef CAS.
- C. T. Chen, B. N. Lin, Y. Y. Hsu, J. D. Liao, W. H. Cheng, C. Y. Lin, H. C. Ku, J. F. Lee, L. Y. Jang and D. G. Liu, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 67, 214424 CrossRef.
- A. V. Gavrikov, P. S. Koroteev, Z. V. Dobrokhotova, A. B. Ilyukhin, N. N. Efimov, D. I. Kirdyankin, M. A. Bykov, M. A. Ryumin and V. M. Novotortsev, Polyhedron, 2015, 102, 48 CrossRef CAS.
- J. H. Van Vleck, J. Appl. Phys., 1968, 39, 365 CrossRef CAS.
- A. Kumar and S. M. Yusuf, Phys. Rep.-Rev. Sec. Phys. Lett., 2015, 556, 1 CAS.
- E. O. Wollan and W. C. Koehler, Phys. Rev., 1955, 100, 545 CrossRef CAS.
- C. Zener, Phys. Rev., 1951, 82, 403 CrossRef CAS.
- K. Yoshii, J. Solid State Chem., 2001, 159, 204 CrossRef CAS.
- L. Néel, Ann. Phys., 1948, 3, 137 Search PubMed.
- K. P. Belov, A. N. Goryaga and T. Y. Gridasova, Fiz. Tverd. Tela, 1972, 14, 1428 CAS.
- T. V. Chandrasekhar Rao, P. Raj, S. M. Yusuf, L. M. Rao, A. Sathyamoorthy and V. C. Sahni, Philos. Mag. B, 1996, 74, 275–291 CrossRef.
- M. Mączka, A. Gągor, B. Macalik, A. Pikul, M. Ptak and J. Hanuza, Inorg. Chem., 2014, 53, 457–467 CrossRef PubMed.
- N. Ihzaz, H. Vincent, G. Dezanneau, H. Roussel, J. Dhahri and M. Oumezzine, J. Magn. Magn. Mater., 2004, 281, 221–226 CrossRef CAS.
- W. S. Ferreira, J. A. Moreira, A. Almeida, M. R. Chaves, J. P. Aráujo, J. B. Oliveira, J. M. M. Da Silva, M. A. Sá, T. M. Mendonça and P. S. Carvalho, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 05430 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra27426e |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 


 ) was 6.24 μB, which is similar to Y. Tadokoro's 5.9 μB
) was 6.24 μB, which is similar to Y. Tadokoro's 5.9 μB